 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ker: Crystal structure of D113A mutant of Drosophila melanogaster Nopp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ker | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of D113A mutant of Drosophila melanogaster Noppera-bo, glutathione S-transferase epsilon 14 (DmGSTE14), in glutathione-bound form | ||||||||||||
 Components Components | Glutathione S-transferase E14 | ||||||||||||
 Keywords Keywords |  TRANSFERASE / Glutathione / Glutathione S-transferase / GST TRANSFERASE / Glutathione / Glutathione S-transferase / GST | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationecdysteroid biosynthetic process / steroid delta-isomerase activity / glutathione transferase / glutathione transferase activity / glutathione metabolic process / cholesterol homeostasis / response to toxic substance / cytoplasmSimilarity search - Function | ||||||||||||
| Biological species |  Drosophila melanogaster (fruit fly) Drosophila melanogaster (fruit fly) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.84 Å | ||||||||||||
 Authors Authors | Koiwai, K. / Inaba, K. / Morohashi, K. / Yumoto, F. / Niwa, R. / Senda, T. | ||||||||||||
| Funding support |  Japan, 3items Japan, 3items
| ||||||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2020 Title: An integrated approach to unravel a crucial structural property required for the function of the insect steroidogenic Halloween protein Noppera-bo. Authors: Koiwai, K. / Inaba, K. / Morohashi, K. / Enya, S. / Arai, R. / Kojima, H. / Okabe, T. / Fujikawa, Y. / Inoue, H. / Yoshino, R. / Hirokawa, T. / Kato, K. / Fukuzawa, K. / Shimada-Niwa, Y. / ...Authors: Koiwai, K. / Inaba, K. / Morohashi, K. / Enya, S. / Arai, R. / Kojima, H. / Okabe, T. / Fujikawa, Y. / Inoue, H. / Yoshino, R. / Hirokawa, T. / Kato, K. / Fukuzawa, K. / Shimada-Niwa, Y. / Nakamura, A. / Yumoto, F. / Senda, T. / Niwa, R. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ker.cif.gz | 128.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ker.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 6ker.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ke/6kerftp://data.pdbj.org/pub/pdb/validation_reports/ke/6ker | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6kelC  6kemSC  6kenC  6keoC  6kepC  6keqC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27499.756 Da / Num. of mol.: 2 / Mutation: D113A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Drosophila melanogaster (fruit fly) / Gene: GstE14, GSTD14-14, nobo, CG4688 / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: Q7JYX0, glutathione transferase Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: Q7JYX0, glutathione transferase#2: Chemical | Glutathione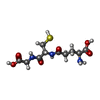  Mass: 307.323 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C10H17N3O6S / Feature type: SUBJECT OF INVESTIGATION Mass: 307.323 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: C10H17N3O6S / Feature type: SUBJECT OF INVESTIGATION#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 146 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Sequence details | Authors state that the conflicts are due to natural valiant. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.33 Å3/Da / Density % sol: 42.33 % / Description: Orthorhombic |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.4 / Details: 42.5%(v/v) PPG400, 100mM Bis-Tris, and pH 6.4 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory / Beamline: BL-1A / Wavelength: 1.1 Å |
| Detector | Type: DECTRIS EIGER X 4M / Detector: PIXEL / Date: Jan 29, 2018 |
| Radiation | Monochromator: Si111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.838→46.34 Å / Num. obs: 41679 / % possible obs: 99.9 % / Redundancy: 13.5 % / Biso Wilson estimate: 21.85 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.113 / Rpim(I) all: 0.0317 / Rrim(I) all: 0.117 / Net I/σ(I): 15.24 |
| Reflection shell | Resolution: 1.838→1.904 Å / Redundancy: 13.9 % / Rmerge(I) obs: 1.124 / Mean I/σ(I) obs: 2.12 / Num. unique obs: 4083 / CC1/2: 0.787 / Rpim(I) all: 0.309 / Rrim(I) all: 1.166 / % possible all: 99.51 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6KEM Resolution: 1.84→46.03 Å / SU ML: 0.1993 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 20.5772
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.04 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.84→46.03 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
