 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5c58: A double mutant of serratia marcescens hemophore receptor HasR in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5c58 | ||||||
|---|---|---|---|---|---|---|---|
| Title | A double mutant of serratia marcescens hemophore receptor HasR in complex with its hemophore HasA and heme | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  TRANSPORT PROTEIN / outer membrane receptor / transporter complex / heme transfer TRANSPORT PROTEIN / outer membrane receptor / transporter complex / heme transfer | ||||||
| Function / homology |  Function and homology information Function and homology informationheme transmembrane transporter activity / cell outer membrane / extracellular region / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Serratia marcescens (bacteria) Serratia marcescens (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.795 Å | ||||||
 Authors Authors | Becker, S. / Diederichs, K. / Welte, W. | ||||||
 Citation Citation | Journal: Eur.Biophys.J. / Year: 2020 Title: Binding of HasA by its transmembrane receptor HasR follows a conformational funnel mechanism. Authors: Exner, T.E. / Becker, S. / Becker, S. / Boniface-Guiraud, A. / Delepelaire, P. / Diederichs, K. / Welte, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5c58.cif.gz | 349.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5c58.ent.gz | 283.2 KB | Display | PDB format |
| PDBx/mmJSON format | 5c58.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c5/5c58ftp://data.pdbj.org/pub/pdb/validation_reports/c5/5c58 | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj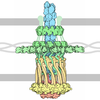
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 94825.602 Da / Num. of mol.: 1 / Mutation: R297A, N800A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Serratia marcescens (bacteria) / Gene: hasR / Production host: Escherichia coli (E. coli) / References: UniProt: Q79AD2 |
|---|---|
| #2: Protein | Mass: 21523.205 Da / Num. of mol.: 1 / Fragment: UNP residues 2-188 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Serratia marcescens (bacteria) / Gene: hasA / Production host: Escherichia coli (E. coli) / References: UniProt: Q54450 |
| #3: Chemical | ChemComp-HEM / Heme B  Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.27 Å3/Da / Density % sol: 62.43 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop Details: 100mM Tris pH 7 to 8, 1.8 to 2.2mM NaCl, 100mM K2HPO4 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Jun 25, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.79→40 Å / Num. obs: 37882 / % possible obs: 99 % / Redundancy: 6.3 % / Rmerge(I) obs: 0.095 / Rsym value: 0.103 / Net I/σ(I): 13.26 |
| Reflection shell | Resolution: 2.79→2.96 Å / Redundancy: 5.9 % / Rmerge(I) obs: 2.227 / Mean I/σ(I) obs: 0.91 / % possible all: 94.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3CSL, 1DK0 Resolution: 2.795→37.749 Å / SU ML: 0.5 / Cross valid method: FREE R-VALUE / σ(F): 1.99 / Phase error: 37.75 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.795→37.749 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
