 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5aab: Structure of C1156Y,L1198F Mutant Human Anaplastic Lymphoma Kinas... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5aab | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of C1156Y,L1198F Mutant Human Anaplastic Lymphoma Kinase in Complex with Crizotinib | ||||||
 Components Components | ALK TYROSINE KINASE RECEPTOR | ||||||
 Keywords Keywords |  TRANSFERASE / RECEPTOR TYROSINE KINASE / ANAPLASTIC LYMPHOMA KINASE / INHIBITOR / MUTANT TRANSFERASE / RECEPTOR TYROSINE KINASE / ANAPLASTIC LYMPHOMA KINASE / INHIBITOR / MUTANT | ||||||
| Function / homology |  Function and homology information Function and homology informationresponse to environmental enrichment / ASP-3026-resistant ALK mutants / NVP-TAE684-resistant ALK mutants / alectinib-resistant ALK mutants / brigatinib-resistant ALK mutants / ceritinib-resistant ALK mutants / crizotinib-resistant ALK mutants / lorlatinib-resistant ALK mutants / receptor signaling protein tyrosine kinase activator activity / regulation of dopamine receptor signaling pathway ...response to environmental enrichment / ASP-3026-resistant ALK mutants / NVP-TAE684-resistant ALK mutants / alectinib-resistant ALK mutants / brigatinib-resistant ALK mutants / ceritinib-resistant ALK mutants / crizotinib-resistant ALK mutants / lorlatinib-resistant ALK mutants / receptor signaling protein tyrosine kinase activator activity / regulation of dopamine receptor signaling pathway / ALK mutants bind TKIs / swimming behavior / positive regulation of dendrite development / regulation of neuron differentiation / adult behavior / Signaling by ALK / Signaling by ALK fusions and activated point mutants / neuron development / Nuclear events stimulated by ALK signaling in cancer / negative regulation of lipid catabolic process / energy homeostasis / peptidyl-tyrosine autophosphorylation / transmembrane receptor protein tyrosine kinase activity / hippocampus development / receptor protein-tyrosine kinase / cell surface receptor protein tyrosine kinase signaling pathway / heparin binding / positive regulation of NF-kappaB transcription factor activity / regulation of cell population proliferation / protein tyrosine kinase activity / regulation of apoptotic process / protein autophosphorylation / receptor complex / phosphorylation / signal transduction / protein-containing complex / extracellular exosome / ATP binding / identical protein binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | McTigue, M. / Deng, Y. / Liu, W. / Brooun, A. / Stewart, A. | ||||||
 Citation Citation | Journal: N.Engl.J.Med. / Year: 2016 Title: Resensitization to Crizotinib by the Lorlatinib Alk Resistance Mutation L1198F. Authors: Shaw, A.T. / Friboulet, L. / Leshchiner, I. / Gainor, J.F. / Bergqvist, S. / Brooun, A. / Burke, B.J. / Deng, Y. / Liu, W. / Dardaei, L. / Frias, R.L. / Schultz, K.R. / Logan, J. / James, L. ...Authors: Shaw, A.T. / Friboulet, L. / Leshchiner, I. / Gainor, J.F. / Bergqvist, S. / Brooun, A. / Burke, B.J. / Deng, Y. / Liu, W. / Dardaei, L. / Frias, R.L. / Schultz, K.R. / Logan, J. / James, L.P. / Smeal, T. / Timofeevski, S. / Katayama, R. / Iafrate, A.J. / Le, L. / Mctigue, M. / Getz, G. / Johnson, T.W. / Engelman, J.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5aab.cif.gz | 77.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5aab.ent.gz | 57.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5aab.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aa/5aabftp://data.pdbj.org/pub/pdb/validation_reports/aa/5aab | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5a9uC  5aa8C  5aa9C  5aaaC  5aacC  2xp2S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37003.402 Da / Num. of mol.: 1 / Fragment: TYROSINE KINASE DOMAIN, UNP RESIDUES 1093-1411 / Mutation: YES Source method: isolated from a genetically manipulated source Details: NONPHOSPHORYLATED / Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PFASTBAC / Cell line (production host): SF9 / Production host:   SPODOPTERA FRUGIPERDA (fall armyworm) SPODOPTERA FRUGIPERDA (fall armyworm)References: UniProt: Q9UM73, receptor protein-tyrosine kinase |
|---|---|
| #2: Chemical | ChemComp-VGH / Crizotinib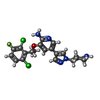  Mass: 450.337 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H22Cl2FN5O / Comment: medication, anticancer, inhibitor*YM Mass: 450.337 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H22Cl2FN5O / Comment: medication, anticancer, inhibitor*YM |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | RESIDUES 1093-1411, CONTAINING MUTATIONS C1156Y AND L1198F, OF HUMAN ANAPLASTIC LYMPHOMA KINASE ...RESIDUES 1093-1411, CONTAINING |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 41 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 286 K / Method: vapor diffusion, hanging drop / pH: 8 Details: HANGING DROP VAPOR DIFFUSION AT 13 DEGREES C. 2 MICROLITERS OF PURIFIED PROTEIN SOLUTION (11-15 MG/ML) CONTAINING INHIBITOR COMPOUND AT A 2X STOICHIOMETRY OF INHIBITOR TO PROTEIN WERE ...Details: HANGING DROP VAPOR DIFFUSION AT 13 DEGREES C. 2 MICROLITERS OF PURIFIED PROTEIN SOLUTION (11-15 MG/ML) CONTAINING INHIBITOR COMPOUND AT A 2X STOICHIOMETRY OF INHIBITOR TO PROTEIN WERE COMBINED WITH 2 MICROLITERS OF SOLUTIONS CONTAINING: 0.2 M LITHIUM SULFATE, 17-21% PEG3350 AND 0.1M TRIS PH 7.6-8.5. |
-Data collection
| Diffraction | Mean temperature: 87 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 0.9795 / Beamline: 17-ID / Wavelength: 0.9795 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 24, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→104.7 Å / Num. obs: 16473 / % possible obs: 99.9 % / Observed criterion σ(I): 1 / Redundancy: 6.4 % / Biso Wilson estimate: 36.5 Å2 / Rmerge(I) obs: 0.11 / Net I/σ(I): 12.7 |
| Reflection shell | Resolution: 2.2→2.46 Å / Redundancy: 6.4 % / Rmerge(I) obs: 0.63 / Mean I/σ(I) obs: 3 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2XP2 Resolution: 2.2→52.39 Å / Rfactor Rfree error: 0.011 / Data cutoff high absF: 1126164.11 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 47.923 Å2 / ksol: 0.369765 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→52.39 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: NONE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.34 Å / Rfactor Rfree error: 0.042 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
