 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4u7h | ||||||
|---|---|---|---|---|---|---|---|
| Title | Oxidized quinone reductase 2 in complex with CK2 inhibitor DMAT | ||||||
 Components Components | Ribosyldihydronicotinamide dehydrogenase [quinone] | ||||||
 Keywords Keywords | Oxidoreductase/Inhibitor / reduced quinone reductase 2 /  DMAT / Oxidoreductase-Inhibitor complex DMAT / Oxidoreductase-Inhibitor complex | ||||||
| Function / homology |  Function and homology informationribosyldihydronicotinamide dehydrogenase (quinone) / dihydronicotinamide riboside quinone reductase activity / quinone catabolic process / resveratrol binding / oxidoreductase activity, acting on other nitrogenous compounds as donors / melatonin binding / NAD(P)H dehydrogenase (quinone) activity / Phase I - Functionalization of compounds / chloride ion binding / FAD binding ...ribosyldihydronicotinamide dehydrogenase (quinone) / dihydronicotinamide riboside quinone reductase activity / quinone catabolic process / resveratrol binding / oxidoreductase activity, acting on other nitrogenous compounds as donors / melatonin binding / NAD(P)H dehydrogenase (quinone) activity / Phase I - Functionalization of compounds / chloride ion binding / FAD binding / electron transfer activity / oxidoreductase activity / protein homodimerization activity / extracellular exosome / zinc ion binding / nucleoplasm / cytosol Function and homology informationribosyldihydronicotinamide dehydrogenase (quinone) / dihydronicotinamide riboside quinone reductase activity / quinone catabolic process / resveratrol binding / oxidoreductase activity, acting on other nitrogenous compounds as donors / melatonin binding / NAD(P)H dehydrogenase (quinone) activity / Phase I - Functionalization of compounds / chloride ion binding / FAD binding ...ribosyldihydronicotinamide dehydrogenase (quinone) / dihydronicotinamide riboside quinone reductase activity / quinone catabolic process / resveratrol binding / oxidoreductase activity, acting on other nitrogenous compounds as donors / melatonin binding / NAD(P)H dehydrogenase (quinone) activity / Phase I - Functionalization of compounds / chloride ion binding / FAD binding / electron transfer activity / oxidoreductase activity / protein homodimerization activity / extracellular exosome / zinc ion binding / nucleoplasm / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.48 Å | ||||||
| Model details | Metallo-flavoprotein | ||||||
 Authors Authors | Leung, K.K. / Shilton, B.H. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2015 Title: Quinone Reductase 2 Is an Adventitious Target of Protein Kinase CK2 Inhibitors TBBz (TBI) and DMAT. Authors: Leung, K.K. / Shilton, B.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4u7h.cif.gz | 197.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4u7h.ent.gz | 157.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4u7h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/u7/4u7hftp://data.pdbj.org/pub/pdb/validation_reports/u7/4u7h | HTTPS FTP |
|---|
-Related structure data





















-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological unit is a dimer and has two active sites. The inhibitor binds NQO2 in two orientations in both active sites. |
-Components
| #1: Protein | Mass: 25849.338 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: NQO2, NMOR2 / Plasmid: pProEXhta / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P16083, EC: 1.10.99.2 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P16083, EC: 1.10.99.2#2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | Flavin adenine dinucleotide  Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#4: Chemical | 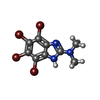  Mass: 476.788 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H7Br4N3 Mass: 476.788 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H7Br4N3#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 551 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 551 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.84 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7 / Details: 1.7M Ammonium sulfate, 0.1M Hepes pH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08B1-1 / Wavelength: 1.0332 Å / Beamline: 08B1-1 / Wavelength: 1.0332 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: May 19, 2011 / Details: 16 CCDs, 16 tiled fiber-optic tapers | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: KOHZU double crystal monochromator (DCM) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.0332 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.45→30.017 Å / Num. obs: 85781 / % possible obs: 96 % / Redundancy: 4.6 % / Biso Wilson estimate: 15.64 Å2 / Rpim(I) all: 0.035 / Rrim(I) all: 0.08 / Rsym value: 0.063 / Net I/av σ(I): 7.996 / Net I/σ(I): 9.7 / Num. measured all: 395937 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.48→30.017 Å / SU ML: 0.14 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 23.15 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 71.86 Å2 / Biso mean: 24.3113 Å2 / Biso min: 9.09 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.48→30.017 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 15
|
