 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ne3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human MHF1-MHF2 complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  DNA BINDING PROTEIN / Histone fold / DNA repair / genome maintenance / Fanconi Anemia / FancM DNA BINDING PROTEIN / Histone fold / DNA repair / genome maintenance / Fanconi Anemia / FancM | ||||||
| Function / homology |  Function and homology information Function and homology informationFANCM-MHF complex / Fanconi anaemia nuclear complex / inner kinetochore / resolution of meiotic recombination intermediates / kinetochore assembly / replication fork processing / Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal / interstrand cross-link repair / Mitotic Prometaphase / EML4 and NUDC in mitotic spindle formation ...FANCM-MHF complex / Fanconi anaemia nuclear complex / inner kinetochore / resolution of meiotic recombination intermediates / kinetochore assembly / replication fork processing / Amplification of signal from unattached kinetochores via a MAD2 inhibitory signal / interstrand cross-link repair / Mitotic Prometaphase / EML4 and NUDC in mitotic spindle formation / Deposition of new CENPA-containing nucleosomes at the centromere / Resolution of Sister Chromatid Cohesion / positive regulation of protein ubiquitination / chromosome segregation / RHO GTPases Activate Formins / Fanconi Anemia Pathway / PKR-mediated signaling / Separation of Sister Chromatids / protein heterodimerization activity / cell division / DNA repair / DNA damage response / chromatin binding / chromatin / DNA binding / nucleoplasm / nucleus / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8007 Å | ||||||
 Authors Authors | Zhao, Q. / Saro, D. / Sachpatzidis, A. / Sung, P. / Xiong, Y. | ||||||
 Citation Citation | Journal: Nat Commun / Year: 2014 Title: The MHF complex senses branched DNA by binding a pair of crossover DNA duplexes. Authors: Zhao, Q. / Saro, D. / Sachpatzidis, A. / Singh, T.R. / Schlingman, D. / Zheng, X.F. / Mack, A. / Tsai, M.S. / Mochrie, S. / Regan, L. / Meetei, A.R. / Sung, P. / Xiong, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ne3.cif.gz | 42.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ne3.ent.gz | 32.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4ne3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ne/4ne3ftp://data.pdbj.org/pub/pdb/validation_reports/ne/4ne3 | HTTPS FTP |
|---|
-Related structure data














-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 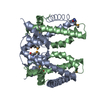
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / CENP-S / Apoptosis-inducing TAF9-like domain-containing protein 1 / FANCM-interacting histone fold ...CENP-S / Apoptosis-inducing TAF9-like domain-containing protein 1 / FANCM-interacting histone fold protein 1 / Fanconi anemia-associated polypeptide of 16 kDa Mass: 10739.987 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: APITD1, CENPS, FAAP16, MHF1 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21 Rossetta / References: UniProt: Q8N2Z9 Escherichia coli (E. coli) / Strain (production host): BL21 Rossetta / References: UniProt: Q8N2Z9 |
|---|---|
| #2: Protein | / CENP-X / FANCM-interacting histone fold protein 2 / Fanconi anemia-associated polypeptide of 10 kDa ...CENP-X / FANCM-interacting histone fold protein 2 / Fanconi anemia-associated polypeptide of 10 kDa / Retinoic acid-inducible gene D9 protein homolog / Stimulated by retinoic acid gene 13 protein homolog Mass: 8445.690 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: STRA13, CENPX, FAAP10, MHF2 / Production host: Escherichia coli (E. coli) / Strain (production host): BL21 Rossetta / References: UniProt: A8MT69 |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.38 % |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 1 Å / Beamline: 24-ID-C / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8007→28.102 Å / Num. all: 16522 / Num. obs: 16358 / % possible obs: 99.01 % / Observed criterion σ(F): 0 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.8007→28.102 Å / Occupancy max: 1 / Occupancy min: 0.47 / FOM work R set: 0.8007 / SU ML: 0.24 / σ(F): 0 / Phase error: 27.05 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 1.06 Å / VDW probe radii: 1.3 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 61.375 Å2 / ksol: 0.409 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 92.75 Å2 / Biso mean: 39.9791 Å2 / Biso min: 18.14 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8007→28.102 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 6
|
