Mass: 35890.828 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Details: RESIDUES 3-32 AT THE N-TERMINUS AND RESIDUES 244-271 OF THE THIRD INTRACELLULAR LOOP WERE DELETED FROM THE CONSTRUCT. THE CONSTRUCT WAS TRUNCATED AFTER RESIDUE 367 AND A HEXAHIS TAG ADDED. Source: (gene. exp.) MELEAGRIS GALLOPAVO (turkey) / Cell: ERYTHROCYTE / Plasmid: PBACPAK8 / Cell line (production host): High Five / Production host: TRICHOPLUSIA NI (cabbage looper) / References: UniProt: P07700
Mass: 18.015 Da / Num. of mol.: 38 / Source method: isolated from a natural source / Formula: H2O
-
Details
Sequence details
THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTABILITY ...THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE THERMOSTABILITY R68S,M90V,I129V,Y227A,A282L,D322K,F327A, F338M,Y343L. THE FOLLOWING MUTATIONS WERE MADE TO IMPROVE EXPRESSION AND HELP CRYSTALLISATION C116L, C358A
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.19 Å3/Da / Density % sol: 44 % / Description: NONE
Crystal grow
Temperature: 293 K / Method: lipidic cubic phase / pH: 7 Details: 25% PEG 600, 0.1M ADA PH7.0 LIPID CUBIC PHASE (LCP) TEMPERATURE 293K
Resolution: 2.1→30.88 Å / Cor.coef. Fo:Fc: 0.939 / Cor.coef. Fo:Fc free: 0.904 / SU B: 4.814 / SU ML: 0.131 / Cross valid method: THROUGHOUT / ESU R: 0.238 / ESU R Free: 0.199 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.24552
964
5.2 %
RANDOM
Rwork
0.19287
-
-
-
obs
0.19558
17696
97.5 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components
 Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj









 Assembly
Assembly




 Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na
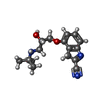
Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 287.357 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H21N3O2
Mass: 287.357 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H21N3O2 Mass: 190.154 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10N2O5 / Comment: pH buffer*YM
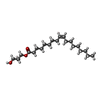
Mass: 190.154 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10N2O5 / Comment: pH buffer*YM Mass: 356.540 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C21H40O4
Mass: 356.540 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C21H40O4 Sample preparation
Sample preparation / Beamline: ID23-2 / Wavelength: 0.8726
/ Beamline: ID23-2 / Wavelength: 0.8726  Processing
Processing