 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4bal: Thaumatin from Thaumatococcus daniellii structure in complex with... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4bal | ||||||
|---|---|---|---|---|---|---|---|
| Title | Thaumatin from Thaumatococcus daniellii structure in complex with the europium tris-hydroxymethyltriazoledipicolinate complex at 1.30 A resolution. | ||||||
 Components Components | THAUMATIN-1 | ||||||
 Keywords Keywords | PLANT PROTEIN / CLICK-CHEMISTRY / ANOMALOUS SCATTERING / DE NOVO PHASING / EXPERIMENTAL PHASING / DIPICOLINATE | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  THAUMATOCOCCUS DANIELLII (katemfe) THAUMATOCOCCUS DANIELLII (katemfe) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.298 Å | ||||||
 Authors Authors | Talon, R. / Kahn, R. / Gautier, A. / Nauton, L. / Girard, E. | ||||||
 Citation Citation | Journal: Chem.Commun.(Camb.) / Year: 2012 Title: Clicked Europium Dipicolinate Complexes for Protein X-Ray Structure Determination. Authors: Talon, R. / Nauton, L. / Canet, J.-L. / Kahn, R. / Girard, E. / Gautier, A. #1: Journal: Angew.Chem.Int.Ed.Engl. / Year: 2008Title: Protein Crystallography Through Supramolecular Interactions between a Lanthanide Complex and Arginine. Authors: Pompidor, G. / D'Aleo, A. / Vicat, J. / Toupet, L. / Giraud, N. / Kahn, R. / Maury, O. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2010Title: A Dipicolinate Lanthanide Complex for Solving Protein Structures Using Anomalous Diffraction. Authors: Pompidor, G. / Maury, O. / Vicat, J. / Kahn, R. #3: Journal: Acta Crystallogr.,Sect.D / Year: 2002Title: Gd-Hpdo3A, a Complex to Obtain High-Phasing-Power Heavy-Atom Derivatives for Sad and MAD Experiments: Results with Tetragonal Hen Egg-White Lysozyme. Authors: Girard, E. / Chantalat, L. / Vicat, J. / Kahn, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4bal.cif.gz | 114.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4bal.ent.gz | 87.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4bal.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ba/4balftp://data.pdbj.org/pub/pdb/validation_reports/ba/4bal | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4badC  4bafC  4bapC  4barSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





































-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / THAUMATIN I / TDTHAU Mass: 22227.059 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: SIGMA-ALDRICH POWDER, CATALOG REFERENCE T7638 / Source: (natural) THAUMATOCOCCUS DANIELLII (katemfe) / References: UniProt: P02883 | ||||
|---|---|---|---|---|---|
| #2: Chemical | 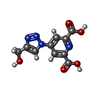  Mass: 264.194 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H8N4O5 Mass: 264.194 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H8N4O5#3: Chemical | ChemComp-EU3 / | Europium  Mass: 151.964 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Eu Mass: 151.964 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Eu#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 376 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 376 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 293 K / pH: 6.5 Details: 0.1 M BIS-TRIS PROPANE PH 6.5, 0.3-0.9 SODIUM POTASSIUM TARTRATE, 0.0006 M PROTEIN, 0.010 M LANTHANIDE COMPLEX, 293 K, 5 DAYS |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM30A / Wavelength: 0.979 / Beamline: BM30A / Wavelength: 0.979 |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Nov 27, 2010 / Details: MIRRORS |
| Radiation | Monochromator: EITHER 111 OR 311 SILICON SINGLE CRISTALS / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→45.9 Å / Num. obs: 64239 / % possible obs: 99.9 % / Observed criterion σ(I): 3 / Redundancy: 6.6 % / Biso Wilson estimate: 10 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 12.5 |
| Reflection shell | Resolution: 1.3→1.37 Å / Redundancy: 6.5 % / Rmerge(I) obs: 0.45 / Mean I/σ(I) obs: 1.7 / % possible all: 99.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4BAR Resolution: 1.298→28.994 Å / SU ML: 0.26 / σ(F): 1.34 / Phase error: 11.87 / Stereochemistry target values: ML Details: ACCORDING TO THE HIGH RESOLUTION, ANISOTROPIC ADP REFINEMENT WAS APPLIED FOR ALL ATOMS. LIGANDS OCCUPANCIES WERE FIXED ACCORDING TO THE LANTHANIDE ION OCCUPANCY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.72 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 47.309 Å2 / ksol: 0.399 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 11.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.298→28.994 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
