Entry Database : PDB / ID : 3zduTitle Crystal structure of the human CDKL3 kinase domain CYCLIN-DEPENDENT KINASE-LIKE 3 Keywords / Function / homology Biological species HOMO SAPIENS (human)Method / / / Resolution : 2.2 Å Authors Canning, P. / Elkins, J.M. / Goubin, S. / Mahajan, P. / Pike, A.C.W. / Quigley, A. / MacKenzie, A. / Carpenter, E.P. / von Delft, F. / Arrowsmith, C.H. ...Canning, P. / Elkins, J.M. / Goubin, S. / Mahajan, P. / Pike, A.C.W. / Quigley, A. / MacKenzie, A. / Carpenter, E.P. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Bullock, A. Journal : Cell Rep / Year : 2018Title : CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function.Authors : Canning, P. / Park, K. / Goncalves, J. / Li, C. / Howard, C.J. / Sharpe, T.D. / Holt, L.J. / Pelletier, L. / Bullock, A.N. / Leroux, M.R. History Deposition Nov 30, 2012 Deposition site / Processing site Revision 1.0 Mar 20, 2013 Provider / Type Revision 1.1 Jan 24, 2018 Group / Structure summary / Category / citation_author / Item / _citation_author.nameRevision 1.2 Mar 30, 2022 Group / Derived calculations / OtherCategory citation / citation_author ... citation / citation_author / database_2 / pdbx_database_status / pdbx_struct_conn_angle / struct_conn / struct_site Item _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr1_symmetry / _pdbx_struct_conn_angle.ptnr2_auth_comp_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr2_label_atom_id / _pdbx_struct_conn_angle.ptnr2_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.ptnr3_symmetry / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr1_symmetry / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn.ptnr2_symmetry / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.3 Dec 20, 2023 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_model
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords TRANSFERASE / PHOSPHO-MIMETIC
TRANSFERASE / PHOSPHO-MIMETIC Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj
 Assembly
Assembly



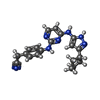




 Mass: 359.428 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H21N7
Mass: 359.428 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H21N7 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Sample preparation
Sample preparation / Beamline: I04 / Wavelength: 0.9611
/ Beamline: I04 / Wavelength: 0.9611  Processing
Processing