 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3vir | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal strcture of Swi5 from fission yeast | ||||||
 Components Components | Mating-type switching protein swi5 | ||||||
 Keywords Keywords | RECOMBINATION ACTIVATOR / Sfr1 | ||||||
| Function / homology |  Function and homology information Function and homology informationmeiotic strand invasion / meiotic DNA recombinase assembly involved in reciprocal meiotic recombination /  Swi5-Sfr1 complex / Swi5-Swi2 complex / meiotic joint molecule formation / gene conversion at mating-type locus / mating type switching / DNA recombinase assembly / DNA strand invasion / ATPase activator activity ...meiotic strand invasion / meiotic DNA recombinase assembly involved in reciprocal meiotic recombination / Swi5-Sfr1 complex / Swi5-Swi2 complex / meiotic joint molecule formation / gene conversion at mating-type locus / mating type switching / DNA recombinase assembly / DNA strand invasion / ATPase activator activity / double-strand break repair via homologous recombination / DNA recombination / identical protein binding Swi5-Sfr1 complex / Swi5-Swi2 complex / meiotic joint molecule formation / gene conversion at mating-type locus / mating type switching / DNA recombinase assembly / DNA strand invasion / ATPase activator activity ...meiotic strand invasion / meiotic DNA recombinase assembly involved in reciprocal meiotic recombination / Swi5-Sfr1 complex / Swi5-Swi2 complex / meiotic joint molecule formation / gene conversion at mating-type locus / mating type switching / DNA recombinase assembly / DNA strand invasion / ATPase activator activity / double-strand break repair via homologous recombination / DNA recombination / identical protein bindingSimilarity search - Function | ||||||
| Biological species |  Schizosaccharomyces pombe (fission yeast) Schizosaccharomyces pombe (fission yeast) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 2.7 Å | ||||||
 Authors Authors | Kuwabara, N. / Yamada, N. / Hashimoto, H. / Sato, M. / Iwasaki, H. / Shimizu, T. | ||||||
 Citation Citation | Journal: Structure / Year: 2012 Title: Mechanistic insights into the activation of Rad51-mediated strand exchange from the structure of a recombination activator, the Swi5-Sfr1 complex Authors: Kuwabara, N. / Murayama, Y. / Hashimoto, H. / Kokabu, Y. / Ikeguchi, M. / Sato, M. / Mayanagi, K. / Tsutsui, Y. / Iwasaki, H. / Shimizu, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3vir.cif.gz | 111.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3vir.ent.gz | 88.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3vir.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/vi/3virftp://data.pdbj.org/pub/pdb/validation_reports/vi/3vir | HTTPS FTP |
|---|
-Related structure data






-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 9764.057 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Schizosaccharomyces pombe (fission yeast)Strain: 972h- / Gene: swi5 / Production host:  Escherichia coli (E. coli) / References: UniProt: Q9UUB7 Escherichia coli (E. coli) / References: UniProt: Q9UUB7#2: Sugar | Octyl glucoside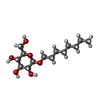  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 2 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.2 Å3/Da / Density % sol: 70.74 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 20% MPD, 0.1M Tris-HCl, 24.5mM octyl-b-glucoside, pH 8.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: AR-NW12A / Wavelength: 1 Å / Beamline: AR-NW12A / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Oct 20, 2005 |
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→50 Å / Num. all: 18023 / Num. obs: 15572 / % possible obs: 86.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Rmerge(I) obs: 0.058 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 2.7→2.8 Å / Rmerge(I) obs: 0.263 / Mean I/σ(I) obs: 4.5 / % possible all: 62.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 2.7→19.65 Å / Cor.coef. Fo:Fc: 0.918 / Cor.coef. Fo:Fc free: 0.875 / SU B: 25.977 / SU ML: 0.24 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.329 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 65.085 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→19.65 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.769 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
