 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3lth | ||||||
|---|---|---|---|---|---|---|---|
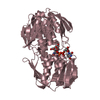
| Title | E. cloacae MurA dead-end complex with UNAG and fosfomycin | ||||||
 Components Components | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | ||||||
 Keywords Keywords | TRANSFERASE/ANTIBIOTIC / open enzyme state / inside-out alpha/beta barrel / Cell wall biogenesis/degradation / Peptidoglycan synthesis / Transferase / TRANSFERASE-ANTIBIOTIC complex | ||||||
| Function / homology |  Function and homology informationUDP-N-acetylglucosamine 1-carboxyvinyltransferase / UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity / UDP-N-acetylgalactosamine biosynthetic process / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / cell cycle / cell division / cytoplasm Function and homology informationUDP-N-acetylglucosamine 1-carboxyvinyltransferase / UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity / UDP-N-acetylgalactosamine biosynthetic process / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / cell cycle / cell division / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Enterobacter cloacae (bacteria) Enterobacter cloacae (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Schonbrunn, E. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2010 Title: The fungal product terreic acid is a covalent inhibitor of the bacterial cell wall biosynthetic enzyme UDP-N-acetylglucosamine 1-carboxyvinyltransferase (MurA) . Authors: Han, H. / Yang, Y. / Olesen, S.H. / Becker, A. / Betzi, S. / Schonbrunn, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3lth.cif.gz | 105.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3lth.ent.gz | 78.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3lth.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lt/3lthftp://data.pdbj.org/pub/pdb/validation_reports/lt/3lth | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3kqaC  3kr6C  1uaeS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / Enoylpyruvate transferase / UDP-N-acetylglucosamine enolpyruvyl transferase / EPT Mass: 44829.406 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacter cloacae (bacteria) / Gene: murA, murZ / Plasmid: pET9d / Production host: Escherichia coli (E. coli) / Strain (production host): BL21References: UniProt: P33038, UDP-N-acetylglucosamine 1-carboxyvinyltransferase |
|---|---|
| #2: Chemical | ChemComp-FFQ / [(Fosfomycin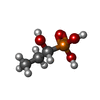  Mass: 140.075 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H9O4P / Comment: antibiotic*YM Mass: 140.075 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H9O4P / Comment: antibiotic*YM |
| #3: Chemical | ChemComp-UD1 /   Mass: 607.354 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H27N3O17P2 Mass: 607.354 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H27N3O17P2 |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 523 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 523 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | THE UNBOUND FORM OF THE ANTIBIOTIC IS FOSFOMYCIN. UPON REACTION WITH PROTEIN, IT COVALENTLY BINDS ...THE UNBOUND FORM OF THE ANTIBIOTIC |
| Sequence details | ASP67 FORMS AN ISOPEPTIDI |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.55 Å3/Da / Density % sol: 51.86 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 6.2 Details: 12.5 mM MES/NaOH (pH 6.2), 25 mM Na/K phosphate buffer, 6% (w/v) polyethylene glycol 20000, VAPOR DIFFUSION, HANGING DROP, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.542 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Oct 4, 1999 / Details: mirrors |
| Radiation | Monochromator: mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.542 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→20 Å / Num. all: 46694 / Num. obs: 46694 / % possible obs: 99.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.8 % / Biso Wilson estimate: 23.5 Å2 / Rmerge(I) obs: 0.084 / Net I/σ(I): 11.7 |
| Reflection shell | Resolution: 1.75→1.79 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 2.2 / Num. unique all: 3050 / % possible all: 98.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1UAE Resolution: 1.75→19.92 Å / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→19.92 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.75→1.86 Å / Rfactor Rfree error: 0.023
|
