 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3hpq: Crystal structure of wild-type adenylate kinase from E. coli, in ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3hpq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of wild-type adenylate kinase from E. coli, in complex with Ap5A | ||||||
 Components Components | Adenylate kinase | ||||||
 Keywords Keywords | TRANSFERASE / Enzyme Inhibitor Complex / ATP-binding / Kinase / Nucleotide biosynthesis / Nucleotide-binding | ||||||
| Function / homology |  Function and homology information Function and homology informationpurine ribonucleotide interconversion / ADP biosynthetic process / nucleoside monophosphate metabolic process / nucleoside diphosphate metabolic process / adenylate kinase / adenylate kinase activity / AMP salvage / nucleoside diphosphate kinase activity / AMP binding / phosphorylation ...purine ribonucleotide interconversion / ADP biosynthetic process / nucleoside monophosphate metabolic process / nucleoside diphosphate metabolic process / adenylate kinase / adenylate kinase activity / AMP salvage / nucleoside diphosphate kinase activity / AMP binding / phosphorylation / magnesium ion binding / ATP binding / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Hilser, V.J. / Travis, T.P. / Bolen, D.W. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2009 Title: Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Authors: Schrank, T.P. / Bolen, D.W. / Hilser, V.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3hpq.cif.gz | 197.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3hpq.ent.gz | 158.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3hpq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hp/3hpqftp://data.pdbj.org/pub/pdb/validation_reports/hp/3hpq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3hprC  1akeS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| 2 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / AK / ATP-AMP transphosphorylase Mass: 23620.029 Da / Num. of mol.: 2 / Mutation: Wild-type Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Strain: K-12 / Gene: adk, b0474, dnaW, JW0463, plsA / Plasmid: pEAK91 / Production host: Escherichia coli (E. coli) / Strain (production host): HMS174 / References: UniProt: P69441, adenylate kinase#2: Chemical | 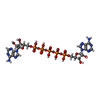  Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5 Mass: 916.367 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H29N10O22P5#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 661 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 661 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.53 Å3/Da / Density % sol: 51.39 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop Details: 45 mg/ml AK in 50 mM MES pH 6.7, 1 mM EDTA, with 50% 50mM MES pH 7.0-7.3, 3% w/v PEG 2000 and 1.8-2.3 Ammonium sulfate, VAPOR DIFFUSION, SITTING DROP, temperature 298K PH range: 7.0-7.3 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: MACSCIENCE / Wavelength: 1.5418 Å |
| Detector | Type: MACSCIENCE / Detector: IMAGE PLATE / Date: Jun 1, 2008 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→50 Å / Num. obs: 36651 / % possible obs: 98.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.9 % / Rmerge(I) obs: 0.046 / Net I/σ(I): 47.55 |
| Reflection shell | Resolution: 1.9→2.03 Å / Rmerge(I) obs: 0.159 / Mean I/σ(I) obs: 9.06 / Num. unique all: 3048 / % possible all: 82.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1AKE Resolution: 2→14.999 Å / SU ML: 0.29 / Isotropic thermal model: anisotropic (tls) / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.8 Å / VDW probe radii: 0.8 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 82.657 Å2 / ksol: 0.381 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.33 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→14.999 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|