- PDB-2wny: Structure of Mth689, a DUF54 protein from Methanothermobacter the... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 2wny
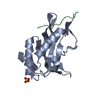
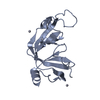
Title
Structure of Mth689, a DUF54 protein from Methanothermobacter thermautotrophicus
Components
CONSERVED PROTEIN MTH689Conservation
Keywords
UNKNOWN FUNCTION / EXOSOME SUPEROPERON
Function / homology
Uncharacterised protein family UPF0201 / RNA binding / 50s Ribosomal Protein L5; Chain: A, / Ribosomal protein L5 / Ribosomal protein L5 domain superfamily / 2-Layer Sandwich / Alpha Beta / NICKEL (II) ION / Conserved protein
Resolution: 1.95→24.23 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.919 / SU B: 9.05 / SU ML: 0.122 / Cross valid method: THROUGHOUT / ESU R: 0.159 / ESU R Free: 0.157 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS. ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.24705
515
2.1 %
RANDOM
Rwork
0.19104
-
-
-
obs
0.19227
23631
99.98 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Conservation
Conservation  Keywords
Keywords Function and homology information
Function and homology information

 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj Assembly
Assembly




 Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni
Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni
 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 158 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.9762
/ Beamline: ID29 / Wavelength: 0.9762  Processing
Processing