 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ieh: Crystal structure of human kinesin Eg5 in complex with (R)-mon97,... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ieh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human kinesin Eg5 in complex with (R)-mon97, a new monastrol-based inhibitor that binds as (R)-enantiomer | ||||||
 Components Components | Kinesin-like protein KIF11 | ||||||
 Keywords Keywords | HYDROLASE / beta-sheet core / flanked by three alpha-helices on each side | ||||||
| Function / homology |  Function and homology information Function and homology informationspindle elongation / Kinesins / plus-end-directed microtubule motor activity / regulation of mitotic centrosome separation / mitotic centrosome separation / COPI-dependent Golgi-to-ER retrograde traffic / kinesin complex / microtubule motor activity / spindle organization / microtubule-based movement ...spindle elongation / Kinesins / plus-end-directed microtubule motor activity / regulation of mitotic centrosome separation / mitotic centrosome separation / COPI-dependent Golgi-to-ER retrograde traffic / kinesin complex / microtubule motor activity / spindle organization / microtubule-based movement / mitotic spindle assembly / MHC class II antigen presentation / mitotic spindle organization / spindle / mitotic spindle / spindle pole / mitotic cell cycle / microtubule binding / microtubule / cell division / protein kinase binding / protein-containing complex / ATP binding / membrane / nucleus / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Garcia-Saez, I. / Kozielski, F. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2007 Title: Structure of human Eg5 in complex with a new monastrol-based inhibitor bound in the R configuration. Authors: Garcia-Saez, I. / DeBonis, S. / Lopez, R. / Trucco, F. / Rousseau, B. / Thuery, P. / Kozielski, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ieh.cif.gz | 154.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ieh.ent.gz | 120.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2ieh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ie/2iehftp://data.pdbj.org/pub/pdb/validation_reports/ie/2ieh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1q0bS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | / Kinesin-related motor protein Eg5 / Kinesin-like spindle protein HKSP / Thyroid receptor- ...Kinesin-related motor protein Eg5 / Kinesin-like spindle protein HKSP / Thyroid receptor-interacting protein 5 / TRIP5 / Kinesin-like protein 1 Mass: 40924.387 Da / Num. of mol.: 2 / Fragment: motor domain of human kinesin Eg5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: KIF11, EG5, KNSL1 / Plasmid: pET28a / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3)pLys / References: UniProt: P52732, EC: 3.6.4.4 Escherichia coli (E. coli) / Strain (production host): BL21(DE3)pLys / References: UniProt: P52732, EC: 3.6.4.4 |
|---|
-Non-polymers , 7 types, 223 molecules 



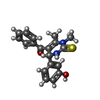


| #2: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-K / |  Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K#4: Chemical | ChemComp-CL / | Chloride Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#5: Chemical | Adenosine diphosphate Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#6: Chemical |  Mass: 338.423 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H18N2O2S Mass: 338.423 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H18N2O2S#7: Chemical | ChemComp-PG4 / | Polyethylene glycol Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#8: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 214 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54.39 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 5.6 Details: 20%PEG4000, 0.2M K2HPO4, 0.1M MES, pH 5.6, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM30A / Wavelength: 0.979645 Å / Beamline: BM30A / Wavelength: 0.979645 Å |
| Detector | Type: CUSTOM-MADE / Detector: CCD / Date: Apr 10, 2006 / Details: mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979645 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→50 Å / Num. all: 52753 / Num. obs: 51576 / % possible obs: 97.8 % / Observed criterion σ(F): 2 / Observed criterion σ(I): 1 / Redundancy: 4 % / Rsym value: 0.19 / Net I/σ(I): 7.71 |
| Reflection shell | Resolution: 2.6→2.7 Å / Redundancy: 2.9 % / Mean I/σ(I) obs: 2.95 / Num. unique all: 2692 / Rsym value: 0.79 / % possible all: 84.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1Q0B Resolution: 2.7→25 Å / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→25 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.87 Å / Rfactor Rfree error: 0.017
|
