 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2i0x | ||||||
|---|---|---|---|---|---|---|---|
| Title | Hypothetical protein PF1117 from Pyrococcus furiosus | ||||||
 Components Components | Hypothetical protein PF1117 Hypothesis Hypothesis | ||||||
 Keywords Keywords | STRUCTURAL GENOMICS / UNKNOWN FUNCTION / PF1117 / HYPOTHETICAL PROTEIN / PYROCOCCUS FURIOSUS / PSI / SOUTHEAST COLLABORATORY FOR STRUCTURAL GENOMICS / Protein Structure Initiative / SECSG | ||||||
| Function / homology |  Function and homology information Function and homology informationmaintenance of CRISPR repeat elements / RNA endonuclease activity / defense response to virus / Hydrolases; Acting on ester bonds / metal ion bindingSimilarity search - Function | ||||||
| Biological species |   Pyrococcus furiosus (archaea) Pyrococcus furiosus (archaea) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.7 Å | ||||||
 Authors Authors | Chen, L.Q. / Fu, Z.-Q. / Liu, Z.-J. / Rose, J.P. / Wang, B.C. / Southeast Collaboratory for Structural Genomics (SECSG) | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of Hypothetical Protein Pf1117 from Pyrococcus furiosus Authors: Chen, L.Q. / Fu, Z.-Q. / Hwang, J. / Chang, J. / Chen, L. / Wang, Y. / Zhang, H. / Liu, Z.-J. / Rose, J.P. / Wang, B.C. / Southeast Collaboratory for Structural Genomics | ||||||
| History |
|
- Structure visualization
Structure visualization
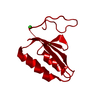
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2i0x.cif.gz | 29.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2i0x.ent.gz | 19.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2i0x.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i0/2i0xftp://data.pdbj.org/pub/pdb/validation_reports/i0/2i0x | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | PISA suggests that the biological assembly is a a2 dimer. The second part of the biological assembly can be generated by the following operator: -Y+2,-X+2,-z+0.5 symmetry ID 8_665 |
-Components
| #1: Protein | Hypothesis Mass: 9992.677 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pyrococcus furiosus (archaea) / Gene: ORF 1117 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3) / References: UniProt: Q8U1T8 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3) / References: UniProt: Q8U1T8 |
|---|---|
| #2: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.45 Å3/Da / Density % sol: 49.77 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 4.2 Details: 1 MICROLITER DROPS CONTAINING EQUAL VOLUMES OF PROTEIN SOLUTION (10 MG/ML) AND A PRECIPITANT SOLUTION CONTAINING 20% PEG 8000, 200 MM SODIUM CHLORIDE, 12% GLYCEROL IN 100MM PHOSPHATE-CITRATE ...Details: 1 MICROLITER DROPS CONTAINING EQUAL VOLUMES OF PROTEIN SOLUTION (10 MG/ML) AND A PRECIPITANT SOLUTION CONTAINING 20% PEG 8000, 200 MM SODIUM CHLORIDE, 12% GLYCEROL IN 100MM PHOSPHATE-CITRATE BUFFER, pH 4.2, VAPOR DIFFUSION, SITTING DROP, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 0.9724 / Wavelength: 0.9724 Å / Beamline: 22-ID / Wavelength: 0.9724 / Wavelength: 0.9724 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Mar 17, 2006 / Details: ROSENBAUM |
| Radiation | Monochromator: SI CHANNEL 220 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9724 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→50 Å / Num. obs: 4033 / % possible obs: 93.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 21.1 % / Rsym value: 0.056 / Net I/σ(I): 20.2 |
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 5.8 % / Mean I/σ(I) obs: 3.7 / Num. unique all: 239 / Rsym value: 0.295 / % possible all: 58.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 2.7→25 Å / Data cutoff high rms absF: 10000 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 46.0341 Å2 / ksol: 0.306465 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 58.2 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→25 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
