 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1y8n | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the PDK3-L2 complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  TRANSFERASE / pyruvate dehydrogenase kinase 3 / lipoyl-bearing domain / protein-protein complex TRANSFERASE / pyruvate dehydrogenase kinase 3 / lipoyl-bearing domain / protein-protein complex | ||||||
| Function / homology |  Function and homology information Function and homology informationhypoxia-inducible factor-1alpha signaling pathway / [pyruvate dehydrogenase (acetyl-transferring)] kinase / regulation of acetyl-CoA biosynthetic process from pyruvate / dihydrolipoyllysine-residue acetyltransferase / dihydrolipoyllysine-residue acetyltransferase activity / acetyl-CoA biosynthetic process from pyruvate / pyruvate dehydrogenase (acetyl-transferring) kinase activity / pyruvate dehydrogenase complex / : / Pyruvate metabolism ...hypoxia-inducible factor-1alpha signaling pathway / [pyruvate dehydrogenase (acetyl-transferring)] kinase / regulation of acetyl-CoA biosynthetic process from pyruvate / dihydrolipoyllysine-residue acetyltransferase / dihydrolipoyllysine-residue acetyltransferase activity / acetyl-CoA biosynthetic process from pyruvate / pyruvate dehydrogenase (acetyl-transferring) kinase activity / pyruvate dehydrogenase complex / : / Pyruvate metabolism / Glyoxylate metabolism and glycine degradation / Regulation of pyruvate dehydrogenase (PDH) complex / regulation of reactive oxygen species metabolic process / cellular response to fatty acid / Signaling by Retinoic Acid / regulation of glucose metabolic process / tricarboxylic acid cycle / peroxisome proliferator activated receptor signaling pathway / cellular response to glucose stimulus / glucose metabolic process / peptidyl-serine phosphorylation / mitochondrial matrix / protein kinase activity / intracellular membrane-bounded organelle / protein serine/threonine kinase activity / nucleolus / mitochondrion / ATP binding / identical protein bindingSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Kato, M. / Chuang, J.L. / Wynn, R.M. / Chuang, D.T. | ||||||
 Citation Citation | Journal: Embo J. / Year: 2005 Title: Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. Authors: Kato, M. / Chuang, J.L. / Tso, S.C. / Wynn, R.M. / Chuang, D.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1y8n.cif.gz | 111.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1y8n.ent.gz | 84.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1y8n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/y8/1y8nftp://data.pdbj.org/pub/pdb/validation_reports/y8/1y8n | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1y8oC  1y8pC  1jm6S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The second part of the biological assembly is generated by the operation: y-x-1, y, 1/2-z. |
-Components
| #1: Protein | [ Mass: 48290.980 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PDK3 / Plasmid: pTrcHisB / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: Q15120, EC: 2.7.1.99 Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: Q15120, EC: 2.7.1.99 |
|---|---|
| #2: Protein | Dihydrolipoyl transacetylase / E2 / Dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase complex / PDC-E2 / 70 ...E2 / Dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase complex / PDC-E2 / 70 kDa mitochondrial autoantigen of primary biliary cirrhosis / PBC / M2 antigen complex 70 kDa subunit Mass: 14192.097 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DLAT, DLTA / Plasmid: pTrcHisB / Species (production host): Escherichia coli / Production host: Escherichia coli BL21 (bacteria) / Strain (production host): BL21References: UniProt: P10515, dihydrolipoyllysine-residue acetyltransferase |
| #3: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K |
| #4: Chemical | ChemComp-RED / Dihydrolipoic acid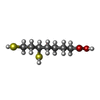  Mass: 208.341 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H16O2S2 Mass: 208.341 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H16O2S2 |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 53 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.7 Å3/Da / Density % sol: 73.7 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.6 Details: soudium citrate, sodium potassium phosphate, sodium chrolide, pH 5.6, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Aug 3, 2004 |
| Radiation | Monochromator: osmic mirror / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→43.69 Å / Num. all: 32454 / Num. obs: 32450 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 9.7 % / Rmerge(I) obs: 0.053 / Net I/σ(I): 28.7 |
| Reflection shell | Resolution: 2.6→2.74 Å / Redundancy: 8.6 % / Rmerge(I) obs: 0.545 / Mean I/σ(I) obs: 3.9 / Num. unique all: 4648 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDK2 PDB entry 1JM6 Resolution: 2.6→43.69 Å / Cor.coef. Fo:Fc: 0.946 / Cor.coef. Fo:Fc free: 0.924 / SU B: 13.189 / SU ML: 0.166 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.278 / ESU R Free: 0.232 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 62.115 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.186 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→43.69 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.667 Å / Total num. of bins used: 20
|
