 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qpe: STRUCTURAL ANALYSIS OF THE LYMPHOCYTE-SPECIFIC KINASE LCK IN COMP... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qpe | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL ANALYSIS OF THE LYMPHOCYTE-SPECIFIC KINASE LCK IN COMPLEX WITH NON-SELECTIVE AND SRC FAMILY SELECTIVE KINASE INHIBITORS | ||||||
 Components Components | LCK KINASE | ||||||
 Keywords Keywords |  TRANSFERASE / ALPHA BETA FOLD TRANSFERASE / ALPHA BETA FOLD | ||||||
| Function / homology |  Function and homology informationregulation of lymphocyte activation / positive regulation of leukocyte cell-cell adhesion / Fc-gamma receptor signaling pathway / CD28 co-stimulation / intracellular zinc ion homeostasis / CD4 receptor binding / FLT3 signaling through SRC family kinases / Nef and signal transduction / Nef Mediated CD4 Down-regulation / Interleukin-2 signaling ...regulation of lymphocyte activation / positive regulation of leukocyte cell-cell adhesion / Fc-gamma receptor signaling pathway / CD28 co-stimulation / intracellular zinc ion homeostasis / CD4 receptor binding / FLT3 signaling through SRC family kinases / Nef and signal transduction / Nef Mediated CD4 Down-regulation / Interleukin-2 signaling / CD28 dependent Vav1 pathway / positive regulation of heterotypic cell-cell adhesion / protein serine/threonine phosphatase activity / Regulation of KIT signaling / CTLA4 inhibitory signaling / phospholipase activator activity / leukocyte migration / positive regulation of T cell receptor signaling pathway / pericentriolar material / Translocation of ZAP-70 to Immunological synapse / Phosphorylation of CD3 and TCR zeta chains / PECAM1 interactions / phospholipase binding / CD28 dependent PI3K/Akt signaling / RHOH GTPase cycle / hemopoiesis / Generation of second messenger molecules / immunological synapse / T cell differentiation / PD-1 signaling / CD8 receptor binding / phosphatidylinositol 3-kinase binding / positive regulation of intrinsic apoptotic signaling pathway / peptidyl-tyrosine autophosphorylation / release of sequestered calcium ion into cytosol / GPVI-mediated activation cascade / T cell costimulation / extrinsic component of cytoplasmic side of plasma membrane / SH2 domain binding / phosphotyrosine residue binding / T cell receptor binding / Signaling by phosphorylated juxtamembrane, extracellular and kinase domain KIT mutants / B cell receptor signaling pathway / non-specific protein-tyrosine kinase / non-membrane spanning protein tyrosine kinase activity / Signaling by SCF-KIT / platelet activation / peptidyl-tyrosine phosphorylation / cell surface receptor protein tyrosine kinase signaling pathway / activation of cysteine-type endopeptidase activity involved in apoptotic process / Constitutive Signaling by Aberrant PI3K in Cancer / positive regulation of T cell activation / Downstream TCR signaling / PIP3 activates AKT signaling / DAP12 signaling / ATPase binding / T cell receptor signaling pathway / PI5P, PP2A and IER3 Regulate PI3K/AKT Signaling / protein phosphatase binding / protein tyrosine kinase activity / intracellular signal transduction / response to xenobiotic stimulus / membrane raft / signaling receptor binding / protein phosphorylation / innate immune response / protein kinase binding / extracellular exosome / ATP binding / identical protein binding / plasma membrane / cytosol Function and homology informationregulation of lymphocyte activation / positive regulation of leukocyte cell-cell adhesion / Fc-gamma receptor signaling pathway / CD28 co-stimulation / intracellular zinc ion homeostasis / CD4 receptor binding / FLT3 signaling through SRC family kinases / Nef and signal transduction / Nef Mediated CD4 Down-regulation / Interleukin-2 signaling ...regulation of lymphocyte activation / positive regulation of leukocyte cell-cell adhesion / Fc-gamma receptor signaling pathway / CD28 co-stimulation / intracellular zinc ion homeostasis / CD4 receptor binding / FLT3 signaling through SRC family kinases / Nef and signal transduction / Nef Mediated CD4 Down-regulation / Interleukin-2 signaling / CD28 dependent Vav1 pathway / positive regulation of heterotypic cell-cell adhesion / protein serine/threonine phosphatase activity / Regulation of KIT signaling / CTLA4 inhibitory signaling / phospholipase activator activity / leukocyte migration / positive regulation of T cell receptor signaling pathway / pericentriolar material / Translocation of ZAP-70 to Immunological synapse / Phosphorylation of CD3 and TCR zeta chains / PECAM1 interactions / phospholipase binding / CD28 dependent PI3K/Akt signaling / RHOH GTPase cycle / hemopoiesis / Generation of second messenger molecules / immunological synapse / T cell differentiation / PD-1 signaling / CD8 receptor binding / phosphatidylinositol 3-kinase binding / positive regulation of intrinsic apoptotic signaling pathway / peptidyl-tyrosine autophosphorylation / release of sequestered calcium ion into cytosol / GPVI-mediated activation cascade / T cell costimulation / extrinsic component of cytoplasmic side of plasma membrane / SH2 domain binding / phosphotyrosine residue binding / T cell receptor binding / Signaling by phosphorylated juxtamembrane, extracellular and kinase domain KIT mutants / B cell receptor signaling pathway / non-specific protein-tyrosine kinase / non-membrane spanning protein tyrosine kinase activity / Signaling by SCF-KIT / platelet activation / peptidyl-tyrosine phosphorylation / cell surface receptor protein tyrosine kinase signaling pathway / activation of cysteine-type endopeptidase activity involved in apoptotic process / Constitutive Signaling by Aberrant PI3K in Cancer / positive regulation of T cell activation / Downstream TCR signaling / PIP3 activates AKT signaling / DAP12 signaling / ATPase binding / T cell receptor signaling pathway / PI5P, PP2A and IER3 Regulate PI3K/AKT Signaling / protein phosphatase binding / protein tyrosine kinase activity / intracellular signal transduction / response to xenobiotic stimulus / membrane raft / signaling receptor binding / protein phosphorylation / innate immune response / protein kinase binding / extracellular exosome / ATP binding / identical protein binding / plasma membrane / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | Zhu, X. / Morgenstern, K.A. | ||||||
 Citation Citation | Journal: Structure Fold.Des. / Year: 1999 Title: Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Authors: Zhu, X. / Kim, J.L. / Newcomb, J.R. / Rose, P.E. / Stover, D.R. / Toledo, L.M. / Zhao, H. / Morgenstern, K.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qpe.cif.gz | 68.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qpe.ent.gz | 54.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1qpe.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qp/1qpeftp://data.pdbj.org/pub/pdb/validation_reports/qp/1qpe | HTTPS FTP |
|---|
-Related structure data























-Links
 PDBj
PDBj















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 32353.818 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: LYMPHOCYTE / Cellular location: CYTOPLASM / Production host:  Trichoplusia ni (cabbage looper) / References: UniProt: P06239, EC: 2.7.1.112 Trichoplusia ni (cabbage looper) / References: UniProt: P06239, EC: 2.7.1.112 | ||||
|---|---|---|---|---|---|
| #2: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-PP2 / | 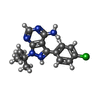  Mass: 302.782 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H17ClN5 Mass: 302.782 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H17ClN5#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.01 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: AMMONIUM SULPHATE, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Feb 7, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→20 Å / Num. obs: 19946 / % possible obs: 93 % / Observed criterion σ(I): -1.5 / Redundancy: 2.7 % / Rmerge(I) obs: 0.082 / Net I/σ(I): 9.4 |
| Reflection shell | Resolution: 2→2.07 Å / Rmerge(I) obs: 0.198 / % possible all: 90 |
| Reflection | *PLUS % possible obs: 93 % / Num. measured all: 53618 |
| Reflection shell | *PLUS % possible obs: 90 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2→5 Å / σ(F): 1 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 98 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 5 Å / σ(F): 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_deg / Dev ideal: 1.8 |
