 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1okg | ||||||
|---|---|---|---|---|---|---|---|
| Title | 3-mercaptopyruvate sulfurtransferase from Leishmania major | ||||||
 Components Components | POSSIBLE 3-MERCAPTOPYRUVATE SULFURTRANSFERASE | ||||||
 Keywords Keywords |  TRANSFERASE / MERCAPTOPYRUVATE / SULFURTRANSFERASE / RHODANESE / PROLYL ISOMERASE / CATALYTIC TRIAD / SERINE PROTEASE / LEISHMANIA PYRUVATE TRANSFERASE / MERCAPTOPYRUVATE / SULFURTRANSFERASE / RHODANESE / PROLYL ISOMERASE / CATALYTIC TRIAD / SERINE PROTEASE / LEISHMANIA PYRUVATE | ||||||
| Function / homology |  Function and homology information3-mercaptopyruvate sulfurtransferase / 3-mercaptopyruvate sulfurtransferase activity / cyanide metabolic process / cysteine biosynthetic process via cystathionine / thiosulfate sulfurtransferase activity / transsulfuration / cytosol Function and homology information3-mercaptopyruvate sulfurtransferase / 3-mercaptopyruvate sulfurtransferase activity / cyanide metabolic process / cysteine biosynthetic process via cystathionine / thiosulfate sulfurtransferase activity / transsulfuration / cytosolSimilarity search - Function | ||||||
| Biological species |  LEISHMANIA MAJOR (eukaryote) LEISHMANIA MAJOR (eukaryote) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 2.1 Å | ||||||
 Authors Authors | Alphey, M.S. / Hunter, W.N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: The Crystal Structure of Leishmania Major 3-Mercaptopyruvate Sulfurtransferase: A Three-Domain Architecture with a Serine Protease-Like Triad at the Active Site Authors: Alphey, M.S. / Williams, R.A.M. / Mottram, J.C. / Coombs, G.H. / Hunter, W.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1okg.cif.gz | 89 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1okg.ent.gz | 72 KB | Display | PDB format |
| PDBx/mmJSON format | 1okg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ok/1okgftp://data.pdbj.org/pub/pdb/validation_reports/ok/1okg | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41297.684 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) LEISHMANIA MAJOR (eukaryote) / Plasmid: PET22A / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): B834 ESCHERICHIA COLI (E. coli) / Strain (production host): B834References: UniProt: Q9NE49, UniProt: Q7K9G0*PLUS, 3-mercaptopyruvate sulfurtransferase | ||
|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca | ||
| #3: Chemical | ChemComp-SO3 / Sulfite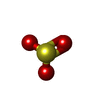  Mass: 80.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO3 Mass: 80.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO3 | ||
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 404 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 404 / Source method: isolated from a natural source / Formula: H2O | ||
| Compound details | ENGINEERED| Sequence details | RESIDUES 371 TO 373 IN THE RECORDS BELOW BELONG TO A SECTION OF THE RECOMBINANT AFFINITY TAG, AND ...RESIDUES 371 TO 373 IN THE RECORDS BELOW BELONG TO A SECTION OF THE RECOMBINAN | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 / Details: pH 8.00 | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jun 21, 2002 / Details: MIRROR |
| Radiation | Monochromator: DIAMOND (111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→30 Å / Num. obs: 23924 / % possible obs: 97.8 % / Redundancy: 5.4 % / Biso Wilson estimate: 36.1 Å2 / Rmerge(I) obs: 0.047 / Net I/σ(I): 23.2 |
| Reflection shell | Resolution: 2.1→2.17 Å / Redundancy: 5.1 % / Rmerge(I) obs: 0.316 / Mean I/σ(I) obs: 4.5 / % possible all: 97.2 |
| Reflection | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 20 Å / Redundancy: 5.4 % / Num. measured all: 131345 / Rmerge(I) obs: 0.047 |
| Reflection shell | *PLUS % possible obs: 97.2 % / Rmerge(I) obs: 0.316 / Mean I/σ(I) obs: 4.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 2.1→76.7 Å / Cor.coef. Fo:Fc: 0.944 / Cor.coef. Fo:Fc free: 0.899 / SU B: 5.558 / SU ML: 0.149 / Cross valid method: THROUGHOUT / ESU R: 0.254 / ESU R Free: 0.232 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: RESIDUES 1-6 AND IN LOOP 239-243 WERE DISORDERED AND COULD NOT BE MODELED. MOST REFINEMENT CARRIED OUT AGAINST A TRUNCATED DATASET TO AVOID RADIATION DAMAGE EFFECTS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44.06 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→76.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|