 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1o9p: Crystal structure of the S131A mutant of Malonamidase E2 complexe... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1o9p | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the S131A mutant of Malonamidase E2 complexed with malonate from Bradyrhizobium japonicum | ||||||
 Components Components | MALONAMIDASE E2 | ||||||
 Keywords Keywords |  HYDROLASE / AMIDASE / MALONATE / MALONAMIDASE HYDROLASE / AMIDASE / MALONATE / MALONAMIDASE | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  BRADYRHIZOBIUM JAPONICUM (bacteria) BRADYRHIZOBIUM JAPONICUM (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / DIRECT METHODS / Resolution: 1.8 Å | ||||||
 Authors Authors | Shin, S. / Oh, B.-H. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: Characterization of a Novel Ser-Cisser-Lys Catalytic Triad in Comparison with the Classical Ser-His-Asp Triad Authors: Shin, S. / Yun, Y.S. / Koo, H.M. / Kim, Y.S. / Choi, K.Y. / Oh, B.-H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1o9p.cif.gz | 171.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1o9p.ent.gz | 135.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1o9p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o9/1o9pftp://data.pdbj.org/pub/pdb/validation_reports/o9/1o9p | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1o9nC  1o9oC  1o9qC  1obiC  1objC  1obkC  1oblC  1ochC  1ockC  1oclC  1ocmC  1gr9 C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 43719.750 Da / Num. of mol.: 2 / Mutation: YES Source method: isolated from a genetically manipulated source Details: MALONATE / Source: (gene. exp.) BRADYRHIZOBIUM JAPONICUM (bacteria) / Plasmid: PUC18 / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: Q9ZIV5#2: Chemical | Malonic acid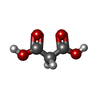  Mass: 104.061 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H4O4 Mass: 104.061 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H4O4#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 564 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 564 / Source method: isolated from a natural source / Formula: H2OCompound details | CIS PEPTIDE BOND BETWEEN GLYCINE 130 AND ALANINE 131 ENGINEERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 39.9 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: 20% POLYETHYLENE GLYCOL 1000, 100 MM TRIS, PH 7.0, 15 MM MALONATE | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU IMAGE PLATE / Detector: IMAGE PLATE / Details: MIRRORS |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.78→20 Å / Num. obs: 71433 / % possible obs: 94.3 % / Observed criterion σ(I): 1 / Redundancy: 4 % / Rsym value: 0.053 / Net I/σ(I): 22.23 |
| Reflection shell | Resolution: 1.78→1.84 Å / Redundancy: 2.8 % / Mean I/σ(I) obs: 4 / Rsym value: 0.198 / % possible all: 84.7 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 20 Å / Num. measured all: 514152 / Rmerge(I) obs: 0.053 |
| Reflection shell | *PLUS % possible obs: 84.7 % / Rmerge(I) obs: 0.198 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIRECT METHODS Starting model: PDB ENTRY 1GR9 1gr9 Resolution: 1.8→20 Å / Cross valid method: THROUGHOUT / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
