 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lra: CRYSTALLOGRAPHIC STUDY OF GLU 58 ALA RNASE T1(ASTERISK)2'-GUANOSI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lra | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTALLOGRAPHIC STUDY OF GLU 58 ALA RNASE T1(ASTERISK)2'-GUANOSINE MONOPHOSPHATE AT 1.9 ANGSTROMS RESOLUTION | ||||||
 Components Components | RIBONUCLEASE T1 | ||||||
 Keywords Keywords | HYDROLASE(ENDORIBONUCLEASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationhyphal tip / ribonuclease T1 activity / ribonuclease T1 / cell septum / endonuclease activity / lyase activity / RNA bindingSimilarity search - Function | ||||||
| Biological species |  Aspergillus oryzae (mold) Aspergillus oryzae (mold) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Pletinckx, J. / Steyaert, J. / Choe, H.-W. / Heinemann, U. / Wyns, L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1994 Title: Crystallographic study of Glu58Ala RNase T1 x 2'-guanosine monophosphate at 1.9-A resolution. Authors: Pletinckx, J. / Steyaert, J. / Zegers, I. / Choe, H.W. / Heinemann, U. / Wyns, L. #1: Journal: J.Mol.Biol. / Year: 1989Title: Three-Dimensional Structure of Ribonuclease T1 Complexed with Guanylyl-2',5'-Guanosine at 1.8 Angstroms Resolution Authors: Koepke, J. / Maslowska, M. / Heinemann, U. / Saenger, W. #2: Journal: J.Biol.Chem. / Year: 1988Title: Three-Dimensional Structure of the Ribonuclease T1(Asterisk)2'-Gmp Complex at 1.9-Angstroms Resolution Authors: Arni, R. / Heinemann, U. / Tokuoka, R. / Saenger, W. #3: Journal: Acta Crystallogr.,Sect.B / Year: 1987Title: Restrained Least-Squares Refinement of the Crystal Structure of the Ribonuclease T1(Asterisk)2'-Guanylic Acid Complex at 1.9 Angstroms Resolution Authors: Arni, R. / Heinemann, U. / Maslowska, M. / Tokuoka, R. / Saenger, W. #4: Journal: Trends Biochem.Sci.(Pers. Ed.) / Year: 1983Title: The Structural and Sequence Homology of a Family of Microbial Ribonucleases Authors: Hill, C. / Dodson, G. / Heinemann, U. / Saenger, W. / Mitsui, Y. / Nakamura, K. / Borisov, S. / Tischenko, G. / Polyakov, K. / Pavlovsky, S. #5: Journal: J.Biomol.Struct.Dyn. / Year: 1983Title: Crystallographic Study of Mechanism of Ribonuclease T1-Catalysed Specific RNA Hydrolysis Authors: Heinemann, U. / Saenger, W. #6: Journal: Nature / Year: 1982Title: Specific Protein-Nucleic Acid Recognition in Ribonuclease T1-2'-Guanylic Acid Complex. An X-Ray Study Authors: Heinemann, U. / Saenger, W. #7: Journal: Eur.J.Biochem. / Year: 1980Title: Crystallization of a Complex between Ribonuclease T1 and 2'-Guanylic Acid Authors: Heinemann, U. / Wernitz, M. / Paehler, A. / Saenger, W. / Menke, G. / Rueterjans, H. #8: Journal: J.Mol.Biol. / Year: 1991Title: Ribonuclease T1 with Free Recognition and Catalytic Site: Crystal Structure Analysis at 1.5 Angstroms Authors: Martinez-Oyanedel, J. / Choe, H.-W. / Heinemann, U. / Saenger, W. #9: Journal: Biochemistry / Year: 1990Title: Histidine-40 of Ribonuclease T1Acts as Base Catalyst When the True Catalytic Base, Glutamic Acid 58 is Replaced by Alanine Authors: Steyaert, J. / Hallenga, K. / Wyns, L. / Stanssens, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lra.cif.gz | 32 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lra.ent.gz | 23.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1lra.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lr/1lraftp://data.pdbj.org/pub/pdb/validation_reports/lr/1lra | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO 39 / 2: CIS PROLINE - PRO 55 |
-Components
| #1: Protein | Mass: 11036.658 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Aspergillus oryzae (mold) / References: UniProt: P00651, EC: 3.1.27.3 |
|---|---|
| #2: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
| #3: Chemical | ChemComp-2GP / Guanosine monophosphate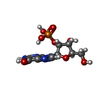  Mass: 363.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O8P Mass: 363.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O8P |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 90 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 90 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.88 Å3/Da / Density % sol: 34.41 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 4.2 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Reflection | *PLUS Highest resolution: 1.9 Å / Rmerge(I) obs: 0.069 |
|---|
- Processing
Processing
| Software | Name: PROFFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→10 Å / σ(F): 1 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROFT / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 10 Å / Num. reflection obs: 5659 / σ(F): 1 / Rfactor obs: 0.178 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
