 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1lr4: Room Temperature Crystal Structure of the Apo-form of the catalyt... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lr4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Room Temperature Crystal Structure of the Apo-form of the catalytic subunit of protein kinase CK2 from Zea mays | |||||||||
 Components Components | Protein kinase CK2 Casein kinase 2 Casein kinase 2 | |||||||||
 Keywords Keywords | TRANSFERASE / protein kinase / CK2 / casein kinase 2 / dual-cosubstrate specificity | |||||||||
| Function / homology |  Function and homology informationprotein kinase CK2 complex / regulation of cell cycle / non-specific serine/threonine protein kinase / phosphorylation / protein serine kinase activity / protein serine/threonine kinase activity / ATP binding / nucleus / cytosol Function and homology informationprotein kinase CK2 complex / regulation of cell cycle / non-specific serine/threonine protein kinase / phosphorylation / protein serine kinase activity / protein serine/threonine kinase activity / ATP binding / nucleus / cytosolSimilarity search - Function | |||||||||
| Biological species |  Zea mays (maize) Zea mays (maize) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | |||||||||
 Authors Authors | Niefind, K. / Puetter, M. / Guerra, B. / Issinger, O.-G. / Schomburg, D. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2005 Title: Inclining the purine base binding plane in protein kinase CK2 by exchanging the flanking side-chains generates a preference for ATP as a cosubstrate. Authors: Yde, C.W. / Ermakova, I. / Issinger, O.G. / Niefind, K. #1: Journal: Embo J. / Year: 1998 Title: Crystal structure of the catalytic subunit of protein kinase CK2 from Zea mays at 2.1 A resolution Authors: Niefind, K. / Guerra, B. / Pinna, L.A. / Issinger, O.-G. / Schomburg, D. #2: Journal: Acta Crystallogr.,Sect.D / Year: 1998 Title: Expression, purification and crystallization of the catalytic subunit of protein kinase CK2 from Zea mays Authors: Guerra, B. / Niefind, K. / Pinna, L.A. / Schomburg, D. / Issinger, O.-G. #3: Journal: NAT.STRUCT.BIOL. / Year: 1999Title: GTP plus water mimic ATP in the active site of protein kinase CK2 Authors: Niefind, K. / Putter, M. / Guerra, B. / Issinger, O.-G. / Schomburg, D. #4: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Crystallization and preliminary characterization of crystals of human protein kinase CK2 Authors: Niefind, K. / Guerra, B. / Ermakowa, I. / Issinger, O.-G. #5: Journal: Embo J. / Year: 2001Title: Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme Authors: Niefind, K. / Guerra, B. / Ermakowa, I. / Issinger, O.-G. #6: Journal: BIOCHIM.BIOPHYS.ACTA / Year: 1993 Title: Expression and characterization of a recombinant maize CK-2 alpha subunit Authors: BOLDYREFF, B. / MEGGIO, F. / DOBROWOLSKA, G. / PINNA, L.A. / Issinger, O.-G. #7: Journal: BIOCHIM.BIOPHYS.ACTA / Year: 1991 Title: Cloning and sequencing of the casein kinase 2 alpha subunit from Zea mays Authors: DOBROWOLSKA, G. / BOLDYREFF, B. / Issinger, O.-G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lr4.cif.gz | 88.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lr4.ent.gz | 66.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1lr4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lr/1lr4ftp://data.pdbj.org/pub/pdb/validation_reports/lr/1lr4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1lp4C  1lpuC  3juhC  1hclS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |























-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Casein kinase 2 / Casein kinase II / alpha chain Mass: 39291.164 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Zea mays (maize) / Plasmid: pt7-7 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3) / References: UniProt: P28523, EC: 2.7.1.37 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3) / References: UniProt: P28523, EC: 2.7.1.37 |
|---|---|
| #2: Chemical | ChemComp-BEN / Benzamidine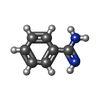  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 225 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 225 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 6 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.46 Å3/Da / Density % sol: 50.09 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8 Details: PEG4000, sodium acetate, Tris/HCl, pH 8.0, VAPOR DIFFUSION, SITTING DROP, temperature 293K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 8.1 / Details: used microseeding | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1.1 Å / Beamline: BW6 / Wavelength: 1.1 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 2, 1996 / Details: Mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→26.2 Å / Num. all: 25896 / Num. obs: 24342 / % possible obs: 94 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.8 % / Biso Wilson estimate: 15.2 Å2 / Rsym value: 0.106 |
| Reflection shell | Resolution: 2→2.11 Å / Redundancy: 1.7 % / Rsym value: 0.111 / % possible all: 84.9 |
| Reflection | *PLUS Lowest resolution: 26 Å / % possible obs: 93.7 % / Rmerge(I) obs: 0.106 |
| Reflection shell | *PLUS Highest resolution: 2 Å / % possible obs: 84.9 % / Rmerge(I) obs: 0.111 / Mean I/σ(I) obs: 4.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: BACKBONE OF CYCLIN-DEPENDENT KINASE 2 (PDB entry 1HCL) Resolution: 2→26 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.927 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.209 / ESU R Free: 0.178 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.212 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→26 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.193 / Rfactor Rfree: 0.237 / Rfactor Rwork: 0.193 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2 Å / Lowest resolution: 2.05 Å / Rfactor obs: 0.216 |
