 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kqr: Crystal Structure of the Rhesus Rotavirus VP4 Sialic Acid Binding... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kqr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Rhesus Rotavirus VP4 Sialic Acid Binding Domain in Complex with 2-O-methyl-alpha-D-N-acetyl neuraminic acid | ||||||
 Components Components | VP4 | ||||||
 Keywords Keywords |  VIRAL PROTEIN / rotavirus / VP4 / VP8* / spike protein / outer capsid / sialic acid / hemagglutinin / cell attachment / neutralization antigen / lectin / galectin fold VIRAL PROTEIN / rotavirus / VP4 / VP8* / spike protein / outer capsid / sialic acid / hemagglutinin / cell attachment / neutralization antigen / lectin / galectin fold | ||||||
| Function / homology |  Function and homology information Function and homology informationhost cell rough endoplasmic reticulum / host cytoskeleton / viral outer capsid / permeabilization of host organelle membrane involved in viral entry into host cell / symbiont entry into host cell via permeabilization of inner membrane / host cell endoplasmic reticulum-Golgi intermediate compartment / virion attachment to host cell / host cell plasma membrane / membraneSimilarity search - Function | ||||||
| Biological species |  Rhesus rotavirus Rhesus rotavirus | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SIR / Resolution: 1.4 Å | ||||||
 Authors Authors | Dormitzer, P.R. / Sun, Z.-Y.J. / Wagner, G. / Harrison, S.C. | ||||||
 Citation Citation | Journal: Embo J. / Year: 2002 Title: The Rhesus Rotavirus VP4 Sialic Acid Binding Domain has a Galectin Fold with a Novel Carbohydrate Binding Site Authors: Dormitzer, P.R. / Sun, Z.-Y.J. / Wagner, G. / Harrison, S.C. #1: Journal: J.Virol. / Year: 2001Title: Proteolysis of Monomeric Recombinant Rotavirus VP4 Yields an Oligomeric VP5* Core Authors: Dormitzer, P.R. / Greenberg, H.B. / Harrison, S.C. | ||||||
| History |
| ||||||
| Remark 12 | The 8 to 16 residue amino terminal linker and residues E62 to V64 of VP4 are disordered and are not ...The 8 to 16 residue amino terminal linker and residues E62 to V64 of VP4 are disordered and are not included in the model. | ||||||
| Remark 13 | Alternate conformations are modeled for residues Q70, M82, I106, S118, T124, Q125, I141, V143, ...Alternate conformations are modeled for residues Q70, M82, I106, S118, T124, Q125, I141, V143, S151, Y155, G156, P157, Q159, K163, V167, N171, N178, E180, K187, E212, S214, and N222. | ||||||
| Remark 14 | The G156-P157 peptide bond has alternate cis and trans conformations. | ||||||
| Remark 15 | The model contains two apparent close contacts: the N222 side chain with HOH 2187 and the G156 ...The model contains two apparent close contacts: the N222 side chain with HOH 2187 and the G156 carbonyl with HOH 2190. In each case, the residue has alternate conformations, and the water molecule has partial occupancy, so that clashes are avoided. | ||||||
| Remark 16 | The electron density for the K187 side chain is ambiguous beyond CB. A well-defined volume of high ...The electron density for the K187 side chain is ambiguous beyond CB. A well-defined volume of high electron density near K187 is modeled with NZ of K187 conformation B. The low B-factor of this atom (2.63) indicates that, in another conformation, this density is occupied by another atom, possibly an ion, which has not been modeled. | ||||||
| Remark 700 | sheet determination method: author |
- Structure visualization
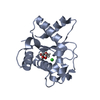
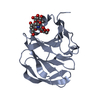
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kqr.cif.gz | 53.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kqr.ent.gz | 37.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1kqr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kq/1kqrftp://data.pdbj.org/pub/pdb/validation_reports/kq/1kqr | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 20220.279 Da / Num. of mol.: 1 / Fragment: sialic acid binding domain (residues 62-224) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rhesus rotavirus / Species: Rotavirus A / Gene: segment 4 / Plasmid: pGex-VP8(62-224) / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 DE3 / References: UniProt: P12473 Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 DE3 / References: UniProt: P12473 |
|---|---|
| #2: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Sugar | ChemComp-MNA / 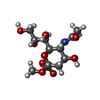  Type: D-saccharide / Mass: 323.296 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H21NO9 Type: D-saccharide / Mass: 323.296 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H21NO9 |
| #4: Chemical | ChemComp-GOL / Glycerol  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 190 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 190 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.84 Å3/Da / Density % sol: 25.2 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 303 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: ammonium sulfate, PEG 400, NaCl, NaPO4, Pipes, Tris, 2-O-methyl-alpha-D-N-acetyl neuraminic acid, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 303K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 14-BM-C / Wavelength: 1 Å / Beamline: 14-BM-C / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Mar 2, 2001 / Details: mirror |
| Radiation | Monochromator: bent G3(III) single crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→30 Å / Num. all: 31002 / Num. obs: 31002 / % possible obs: 99.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 8.25 % / Biso Wilson estimate: 12.8 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 37.4 |
| Reflection shell | Resolution: 1.4→1.43 Å / Redundancy: 5.71 % / Rmerge(I) obs: 0.251 / Mean I/σ(I) obs: 5.51 / Num. unique all: 1993 / % possible all: 98.3 |
| Reflection | *PLUS Highest resolution: 1.4 Å / Lowest resolution: 30 Å / Rmerge(I) obs: 0.05 |
| Reflection shell | *PLUS % possible obs: 98.3 % / Num. unique obs: 1993 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIR / Resolution: 1.4→25 Å / Data cutoff high rms absF: 10000 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.5 Å2 | ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→25 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.4→1.45 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 25 Å / % reflection Rfree: 5 % / Rfactor obs: 0.169 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS % reflection Rfree: 5 % / Rfactor obs: 0.204 |
