 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1k01: Structural Basis for the Interaction of Antibiotics with the Pept... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1k01 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural Basis for the Interaction of Antibiotics with the Peptidyl Transferase Center in Eubacteria | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  RIBOSOME / 50S / 23S / 5S / Antibiotics / Chloramphenicol / Peptidyl transferase center RIBOSOME / 50S / 23S / 5S / Antibiotics / Chloramphenicol / Peptidyl transferase center | ||||||
| Function / homology |  Function and homology information Function and homology informationlarge ribosomal subunit / cytosolic large ribosomal subunit / rRNA binding / ribosome / structural constituent of ribosome / translation / ribonucleoprotein complex / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Deinococcus radiodurans (radioresistant) Deinococcus radiodurans (radioresistant) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 3.5 Å | ||||||
 Authors Authors | Schluenzen, F. / Zarivach, R. / Harms, J. / Bashan, A. / Tocilj, A. / Albrecht, R. / Yonath, A. / Franceschi, F. | ||||||
 Citation Citation | Journal: Nature / Year: 2001 Title: Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Authors: Schlunzen, F. / Zarivach, R. / Harms, J. / Bashan, A. / Tocilj, A. / Albrecht, R. / Yonath, A. / Franceschi, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1k01.cif.gz | 1.3 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1k01.ent.gz | 1 MB | Display | PDB format |
| PDBx/mmJSON format | 1k01.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/k0/1k01ftp://data.pdbj.org/pub/pdb/validation_reports/k0/1k01 | HTTPS FTP |
|---|
-Related structure data
















-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-RNA chain , 1 types, 1 molecules A
| #1: RNA chain | 23S ribosomal RNA Mass: 933405.000 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Deinococcus radiodurans (radioresistant) / References: GenBank: 15805042 |
|---|
-Ribosomal Protein ... , 3 types, 3 molecules KLM
| #2: Protein | 60S ribosomal protein L4 / Coordinate model: Cα atoms only Mass: 22308.156 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Deinococcus radiodurans (radioresistant) / References: UniProt: Q9RXK1 |
|---|---|
| #3: Protein | 60S ribosomal protein L22 / Coordinate model: Cα atoms only Mass: 15190.869 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Deinococcus radiodurans (radioresistant) / References: UniProt: Q9RXJ7 |
| #4: Protein | / 50S ribosomal protein L32 / Coordinate model: Cα atoms only Mass: 6810.017 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Deinococcus radiodurans (radioresistant) / References: UniProt: P49228 |
-Non-polymers , 2 types, 4 molecules 
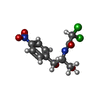
| #5: Chemical |  Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg#6: Chemical | ChemComp-CLM / | Chloramphenicol Mass: 323.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12Cl2N2O5 / Comment: antibiotic*YM Mass: 323.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12Cl2N2O5 / Comment: antibiotic*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4 Å3/Da / Density % sol: 64 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 7.8 Details: ethanol, dimethylhexanediol, MgCl2, KCl, Hepes, NH4Cl, pH 7.8, VAPOR DIFFUSION, HANGING DROP, temperature 291K | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 90 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.9393 Å / Beamline: ID14-4 / Wavelength: 0.9393 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Apr 1, 2001 |
| Radiation | Monochromator: Si111 or Si311 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9393 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→25 Å / Num. all: 238661 / Num. obs: 238661 / % possible obs: 88.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 |
| Reflection shell | Resolution: 3.5→3.62 Å / % possible all: 71 |
| Reflection | *PLUS Lowest resolution: 25 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.124 |
| Reflection shell | *PLUS % possible obs: 71 % / Rmerge(I) obs: 0.577 / Mean I/σ(I) obs: 2.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 3.5→30 Å / σ(F): 0 Details: The coordinates of four chains of the 50s subunit and chloramphenicol were deposited. The number of non-hydrogen atoms used in refinement is more than specified in REMARK 3: 26069 protein ...Details: The coordinates of four chains of the 50s subunit and chloramphenicol were deposited. The number of non-hydrogen atoms used in refinement is more than specified in REMARK 3: 26069 protein atoms, 62115 nucleic acid atoms, and 102 heterogen atoms were used in refinement.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→30 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: CNS/REFMAC / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3.5 Å / Lowest resolution: 30 Å / σ(F): 0 / Rfactor obs: 0.275 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS |
