 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jcn: BINARY COMPLEX OF HUMAN TYPE-I INOSINE MONOPHOSPHATE DEHYDROGENAS... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jcn | ||||||
|---|---|---|---|---|---|---|---|
| Title | BINARY COMPLEX OF HUMAN TYPE-I INOSINE MONOPHOSPHATE DEHYDROGENASE WITH 6-CL-IMP | ||||||
 Components Components | INOSINE MONOPHOSPHATE DEHYDROGENASE I | ||||||
 Keywords Keywords |  OXIDOREDUCTASE / DEHYDROGENASE / IMPD / IMPDH / GUANINE NUCLEOTIDE SYNTHESIS OXIDOREDUCTASE / DEHYDROGENASE / IMPD / IMPDH / GUANINE NUCLEOTIDE SYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationPurine ribonucleoside monophosphate biosynthesis / IMP dehydrogenase activity / IMP dehydrogenase / GMP biosynthetic process / Azathioprine ADME / GTP biosynthetic process / azurophil granule lumen / secretory granule lumen / ficolin-1-rich granule lumen / Potential therapeutics for SARS ...Purine ribonucleoside monophosphate biosynthesis / IMP dehydrogenase activity / IMP dehydrogenase / GMP biosynthetic process / Azathioprine ADME / GTP biosynthetic process / azurophil granule lumen / secretory granule lumen / ficolin-1-rich granule lumen / Potential therapeutics for SARS / nucleic acid binding / nucleotide binding / Neutrophil degranulation / DNA binding / RNA binding / extracellular region / identical protein binding / metal ion binding / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Risal, D. / Strickler, M.D. / Goldstein, B.M. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of the Human Type I Inosine Monophosphate Dehydrogenase and Implications for Isoform Specificity Authors: Risal, D. / Strickler, M.D. / Goldstein, B.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jcn.cif.gz | 168 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jcn.ent.gz | 130.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1jcn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jc/1jcnftp://data.pdbj.org/pub/pdb/validation_reports/jc/1jcn | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.00014, 1, -0.00042), Vector : |
-Components
| #1: Protein | Mass: 55513.613 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: impdh1 / Variant: TYPE I ISOZYME / Plasmid: pIMP1 / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P20839, IMP dehydrogenase Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: P20839, IMP dehydrogenase#2: Chemical | 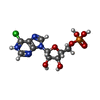  Mass: 367.660 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13ClN4O7P Mass: 367.660 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13ClN4O7P#3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 294 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 294 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 59.17 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: PROTEIN SOLUTION: 7MGS/ML PROTEIN, 50 MM TRIS HCL (PH 8.0), 50 MM KCL, 2MM EDTA, 1MM DTT, 5MM 6-CL-IMP. WELL SOLUTION: 9% (W/V) PEG-800, 100 MM TRIS HCL (PH 8.0), 1.0 M LICL, 24 MM ...Details: PROTEIN SOLUTION: 7MGS/ML PROTEIN, 50 MM TRIS HCL (PH 8.0), 50 MM KCL, 2MM EDTA, 1MM DTT, 5MM 6-CL-IMP. WELL SOLUTION: 9% (W/V) PEG-800, 100 MM TRIS HCL (PH 8.0), 1.0 M LICL, 24 MM BETAMERCAPTOETHANOL (2+2 MICROLITER DROPS), pH 8.00, VAPOR DIFFUSION, HANGING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.91 / Beamline: A1 / Wavelength: 0.91 |
| Detector | Type: CUSTOM-MADE / Detector: CCD / Date: May 5, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→39.74 Å / Num. all: 44411 / Num. obs: 44411 / % possible obs: 97.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3 % / Biso Wilson estimate: 28 Å2 / Rmerge(I) obs: 0.093 / Rsym value: 0.093 / Net I/σ(I): 6.3 |
| Reflection shell | Resolution: 2.5→2.64 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.366 / Mean I/σ(I) obs: 1.9 / Rsym value: 0.366 / % possible all: 93.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: CORE DOMAIN OF IMPDH MONOMER FROM HUMAN TYPE II/6-CL-IMP COMPLEX STRUCTURE Resolution: 2.5→39.74 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 2983561.89 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 Details: 45 RESIDUES OF THE FLANKING DOMAIN AND 50 RESIDUES OF THE ACTIVE-SITE FLAP ARE DISORDERED IN BOTH MONOMERS. 6 RESIDUES IN THE FLANKING DOMAIN HAVE BEEN MODELLED UP TO THE CB DUE TO LACK OF ...Details: 45 RESIDUES OF THE FLANKING DOMAIN AND 50 RESIDUES OF THE ACTIVE-SITE FLAP ARE DISORDERED IN BOTH MONOMERS. 6 RESIDUES IN THE FLANKING DOMAIN HAVE BEEN MODELLED UP TO THE CB DUE TO LACK OF ROBUST SIDE-CHAIN ELECTRON DENSITY. SEGMENTS OF THE CORE DOMAIN RELATED BY NCS-SYMMETRY HAD NCS RESTRAINTS ON BACKBONE C-ALPHA, N, AND C ATOMS DURING REFINEMENT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 48.48 Å2 / ksol: 0.36 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→39.74 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.66 Å / Rfactor Rfree error: 0.012 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
