 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h4w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human trypsin IV (brain trypsin) | ||||||
 Components Components | TRYPSIN IVA | ||||||
 Keywords Keywords |  HYDROLASE / SERINE PROTEASE / SIGNAL TRANSDUCTION / INHIBITOR RESISTANCE / ALZHEIMER DISEASE HYDROLASE / SERINE PROTEASE / SIGNAL TRANSDUCTION / INHIBITOR RESISTANCE / ALZHEIMER DISEASE | ||||||
| Function / homology |  Function and homology information Function and homology informationUptake of dietary cobalamins into enterocytes / antimicrobial humoral response / Alpha-defensins / zymogen activation / Antimicrobial peptides / endothelial cell migration / trypsin / digestion / serine-type peptidase activity / tertiary granule lumen ...Uptake of dietary cobalamins into enterocytes / antimicrobial humoral response / Alpha-defensins / zymogen activation / Antimicrobial peptides / endothelial cell migration / trypsin / digestion / serine-type peptidase activity / tertiary granule lumen / serine-type endopeptidase activity / calcium ion binding / Neutrophil degranulation / proteolysis / extracellular space / extracellular regionSimilarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Katona, G. / Berglund, G.I. / Hajdu, J. / Graf, L. / Szilagyi, L. | ||||||
 Citation Citation | Journal: J. Mol. Biol. / Year: 2002 Title: Crystal structure reveals basis for the inhibitor resistance of human brain trypsin. Authors: Katona, G. / Berglund, G.I. / Hajdu, J. / Graf, L. / Szilagyi, L. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h4w.cif.gz | 61.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h4w.ent.gz | 43.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1h4w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h4/1h4wftp://data.pdbj.org/pub/pdb/validation_reports/h4/1h4w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1trnS S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24301.510 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: BENZAMIDINE / Source: (gene. exp.) HOMO SAPIENS (human) / Organ: BRAIN / Variant: A FORM / Plasmid: PET17B / Production host:  ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P35030, trypsin ESCHERICHIA COLI (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P35030, trypsin |
|---|---|
| #2: Chemical | ChemComp-BEN / Benzamidine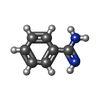  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 |
| #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 125 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | VAL A 86, CLONING ARTIFACT OR NATURAL POLYMORPHI |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.7 Details: HANGING DROP VAPOUR DIFFUSION 3.3 MG/ML PROTEIN, 2% PEG 4000, 0.05M TRIS PH7.7, 2.5MM CACL2, 2.5 MG/ML BENZAMIDINE/HCL, 25 C, pH 7.70 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.886 / Beamline: BM14 / Wavelength: 0.886 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jun 4, 2000 / Details: MIRRORS |
| Radiation | Monochromator: SI 111 CHANNEL CUT / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.886 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→52.7 Å / Num. obs: 26174 / % possible obs: 94.9 % / Redundancy: 4.3 % / Biso Wilson estimate: 15.8 Å2 / Rmerge(I) obs: 0.049 / Net I/σ(I): 18.5 |
| Reflection shell | Resolution: 1.7→1.74 Å / Rmerge(I) obs: 0.323 / Mean I/σ(I) obs: 2.5 / % possible all: 92 |
| Reflection | *PLUS Num. measured all: 111768 |
| Reflection shell | *PLUS % possible obs: 92 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1TRN Resolution: 1.7→52.7 Å / Rfactor Rfree error: 0.006 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 58.8756 Å2 / ksol: 0.359685 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→52.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.76 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 26170 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.23 |
