 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h2y: PROLYL OLIGOPEPTIDASE FROM PORCINE BRAIN, Y473F MUTANT WITH COVAL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h2y | ||||||
|---|---|---|---|---|---|---|---|
| Title | PROLYL OLIGOPEPTIDASE FROM PORCINE BRAIN, Y473F MUTANT WITH COVALENTLY BOUND INHIBITOR Z-PRO-PROLINAL | ||||||
 Components Components | PROLYL ENDOPEPTIDASE | ||||||
 Keywords Keywords | HYDROLASE / PROLYL OLIGOPEPTIDASE / AMNESIA / ALPHA/ BETA- HYDROLASE / BETA-PROPELLER / SERINE PROTEASE | ||||||
| Function / homology |  Function and homology informationprolyl oligopeptidase / oligopeptidase activity / serine-type endopeptidase activity / proteolysis / cytosol Function and homology informationprolyl oligopeptidase / oligopeptidase activity / serine-type endopeptidase activity / proteolysis / cytosolSimilarity search - Function | ||||||
| Biological species |  SUS SCROFA (pig) SUS SCROFA (pig) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.78 Å | ||||||
 Authors Authors | Rea, D. / Fulop, V. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Electrostatic Effects and Binding Determinants in the Catalysis of Prolyl Oligopeptidase: Site Specific Mutagenesis at the Oxyanion Binding Site Authors: Szeltner, Z. / Rea, D. / Renner, V. / Fulop, V. / Polgar, L. #1: Journal: J.Biol.Chem. / Year: 2001Title: Structures of Prolyl Oligopeptidase Substrate/ Inhibitor Complexes. Use of Inhibitor Binding for Titration of the Catalytic Histidine Residue Authors: Fulop, V. / Szeltner, Z. / Renner, V. / Polgar, L. #2: Journal: Embo Rep. / Year: 2000Title: Catalysis of Serine Oligopeptidases is Controlled by a Gating Filter Mechanism Authors: Fulop, V. / Szeltner, Z. / Polgar, L. #3: Journal: Cell(Cambridge,Mass.) / Year: 1998Title: Prolyl Oligopeptidase: An Unusual Beta-Propeller Domain Regulates Proteolysis Authors: Fulop, V. / Bocskei, Z. / Polgar, L. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h2y.cif.gz | 171.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h2y.ent.gz | 134.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1h2y.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h2/1h2yftp://data.pdbj.org/pub/pdb/validation_reports/h2/1h2y | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1h2wC  1h2xC  1h2zC  1qfmS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / POST-PROLINE CLEAVING ENZYME / PE Mass: 80848.344 Da / Num. of mol.: 1 / Mutation: YES Source method: isolated from a genetically manipulated source Details: ENGINEERED MUTATION TYR 473 PHE / Source: (gene. exp.) SUS SCROFA (pig) / Tissue: BRAIN / Production host:  ESCHERICHIA COLI (E. coli) / References: UniProt: P23687, prolyl oligopeptidase ESCHERICHIA COLI (E. coli) / References: UniProt: P23687, prolyl oligopeptidase | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ZPR / 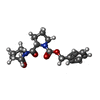  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 330.378 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H22N2O4 / References: Z-PRO-PROLINAL Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 330.378 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H22N2O4 / References: Z-PRO-PROLINAL | ||||
| #3: Chemical | ChemComp-GOL / Glycerol  Mass: 92.094 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 781 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 781 / Source method: isolated from a natural source / Formula: H2OCompound details | CLEAVES PEPTIDE BONDS ON THE C-TERMINAL SIDE OF PROLYL RESIDUES WITHIN PEPTIDES OF UP TO ...CLEAVES PEPTIDE BONDS ON THE C-TERMINAL SIDE OF PROLYL RESIDUES WITHIN PEPTIDES OF UP TO APPROXIMAT | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 44 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.5 / Details: SEE REFERENCE 3, pH 8.50 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop / Details: Fulop, V., (1998) Cell, 94, 161. | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX14.2 / Wavelength: 0.979 / Beamline: PX14.2 / Wavelength: 0.979 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jun 15, 2002 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 1.78→33 Å / Num. obs: 75584 / % possible obs: 99.7 % / Observed criterion σ(I): -3 / Redundancy: 4 % / Biso Wilson estimate: 20.1 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 19.7 |
| Reflection shell | Resolution: 1.78→1.84 Å / Redundancy: 4 % / Rmerge(I) obs: 0.482 / Mean I/σ(I) obs: 2.1 / % possible all: 99.7 |
| Reflection | *PLUS Lowest resolution: 33 Å / Num. measured all: 304631 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1QFM Resolution: 1.78→33 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati sigma a obs: 0.12 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.78→33 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 4 % / Rfactor all: 0.178 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
