 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h16 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Pyruvate Formate-Lyase (E.coli) in complex with Pyruvate and CoA | |||||||||
 Components Components | FORMATE ACETYLTRANSFERASE 1 | |||||||||
 Keywords Keywords |  TRANSFERASE / LYASE / GLYCYL RADICAL ENZYME / ACYLTRANSFERASE TRANSFERASE / LYASE / GLYCYL RADICAL ENZYME / ACYLTRANSFERASE | |||||||||
| Function / homology |  Function and homology informationformate C-acetyltransferase / formate C-acetyltransferase activity / glucose metabolic process / membrane / cytosol Function and homology informationformate C-acetyltransferase / formate C-acetyltransferase activity / glucose metabolic process / membrane / cytosolSimilarity search - Function | |||||||||
| Biological species |  ESCHERICHIA COLI (E. coli) ESCHERICHIA COLI (E. coli) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.53 Å | |||||||||
 Authors Authors | Becker, A. / Kabsch, W. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: X-Ray Structure of Pyruvate Formate-Lyase in Complex with Pyruvate and Coa.How the Enzyme Uses the Cys-418 Thiyl Radical for Pyruvate Cleavage Authors: Becker, A. / Kabsch, W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h16.cif.gz | 204.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h16.ent.gz | 159 KB | Display | PDB format |
| PDBx/mmJSON format | 1h16.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h1/1h16ftp://data.pdbj.org/pub/pdb/validation_reports/h1/1h16 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1h17C  1h18C  3pflS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 85327.898 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: COMPLEX OF PYRUVATE FORMATE-LYASE WITH ITS SUBSTRATES PYRUVATE AND COA Source: (natural) ESCHERICHIA COLI (E. coli) / References: UniProt: P09373, formate C-acetyltransferase |
|---|
-Non-polymers , 7 types, 1282 molecules 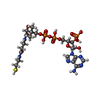





| #2: Chemical | ChemComp-COA / Coenzyme A Mass: 767.534 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H36N7O16P3S Mass: 767.534 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H36N7O16P3S | ||||||||
|---|---|---|---|---|---|---|---|---|---|
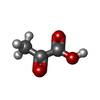
| #3: Chemical | ChemComp-PYR / Pyruvic acid Mass: 88.062 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O3 Mass: 88.062 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H4O3 | ||||||||
| #4: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: Na#5: Chemical | ChemComp-MG / |  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 122.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O4 Mass: 122.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O4#7: Chemical | Polyethylene glycol Mass: 194.226 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#8: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 1267 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Compound details | GLUCOSE NONOXIDATI |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.48 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.3 / Details: pH 7.30 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X13 / Wavelength: 0.8045 / Beamline: X13 / Wavelength: 0.8045 |
| Detector | Type: MARRESEARCH / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8045 Å / Relative weight: 1 |
| Reflection | Resolution: 1.53→15 Å / Num. obs: 128878 / % possible obs: 98.6 % / Redundancy: 8.6 % / Biso Wilson estimate: 16.5 Å2 / Rmerge(I) obs: 0.047 / Net I/σ(I): 28.93 |
| Reflection shell | Resolution: 1.53→1.6 Å / Redundancy: 4.39 % / Rmerge(I) obs: 0.183 / Mean I/σ(I) obs: 7.52 / % possible all: 89.8 |
| Reflection | *PLUS Lowest resolution: 15 Å / Num. measured all: 1104993 / Rmerge(I) obs: 0.042 |
| Reflection shell | *PLUS % possible obs: 89.8 % / Rmerge(I) obs: 0.207 / Mean I/σ(I) obs: 7.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3PFL Resolution: 1.53→15 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 5042753.84 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT Details: ELECTRON DENSITY NEAR PHOSPHATES OF COA WAS EXPLAINED AS MG2+ ALTHOUGH NO DIVALENT CATIONS WERE INCLUDED IN THE CRYSTALLIZATION BUFFER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 52.6765 Å2 / ksol: 0.370898 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.53→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.53→1.63 Å / Rfactor Rfree error: 0.011 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
