 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gpj | ||||||
|---|---|---|---|---|---|---|---|
| Title | Glutamyl-tRNA Reductase from Methanopyrus kandleri | ||||||
 Components Components | Glutamyl-tRNA reductase | ||||||
 Keywords Keywords | REDUCTASE / TRNA-DEPENDENT TETRAPYRROLE BIOSYNTHESIS / GLUTAMYL TRNA- REDUCTASE | ||||||
| Function / homology |  Function and homology informationglutamyl-tRNA reductase / glutamyl-tRNA reductase activity / protoporphyrinogen IX biosynthetic process / NADP binding Function and homology informationglutamyl-tRNA reductase / glutamyl-tRNA reductase activity / protoporphyrinogen IX biosynthetic process / NADP bindingSimilarity search - Function | ||||||
| Biological species |   Methanopyrus kandleri (archaea) Methanopyrus kandleri (archaea) | ||||||
| Method | X-RAY DIFFRACTION / MIR / Resolution: 1.95 Å | ||||||
 Authors Authors | Moser, J. / Schubert, W.-D. / Beier, V. / Bringemeier, I. / Jahn, D. / Heinz, D.W. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 2001 Title: V-shaped structure of glutamyl-tRNA reductase, the first enzyme of tRNA-dependent tetrapyrrole biosynthesis. Authors: Moser, J. / Schubert, W.D. / Beier, V. / Bringemeier, I. / Jahn, D. / Heinz, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gpj.cif.gz | 100.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gpj.ent.gz | 81.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1gpj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gp/1gpjftp://data.pdbj.org/pub/pdb/validation_reports/gp/1gpj | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|

-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | / GluTR Mass: 45439.516 Da / Num. of mol.: 1 / Fragment: WHOLE MOLECULE, RESIDUES 1-404 / Mutation: Cys to Ser Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methanopyrus kandleri (archaea)Description: DSM 6324, GERMAN COLLECTION OF MICROORGANISMS (DSM) Gene: hemA, MK0200 / Production host:  Escherichia coli BL21(DE3) (bacteria) / References: UniProt: Q9UXR8, glutamyl-tRNA reductase Escherichia coli BL21(DE3) (bacteria) / References: UniProt: Q9UXR8, glutamyl-tRNA reductase |
|---|---|
| #2: Chemical | ChemComp-GLU / Glutamic acid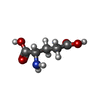  Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO4 Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO4 |
| #3: Chemical | ChemComp-GMC / (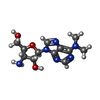  Mass: 294.310 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H18N6O3 Mass: 294.310 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H18N6O3 |
| #4: Chemical | ChemComp-CIT / Citric acid  Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 |
| #5: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 353 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | ALL CYSTEINES REPLACED BY SERINE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.99 Å3/Da / Density % sol: 58.52 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: METHOD: HANGING DROP, TEMP.: 4C, PROTEIN CONCENTRATION: 9.8 MG/ML, PRECIPITANT: 30 MPD, BUFFER: 100MM HEPES PH 7.5, SALT: 200MM NACL, 200MM NACITRATE, 2MM MGCL2 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop / pH: 7.5 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: May 15, 2000 / Details: MIRRORS |
| Radiation | Monochromator: MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.93→30.7 Å / Num. obs: 32681 / % possible obs: 91.1 % / Redundancy: 2.5 % / Rmerge(I) obs: 0.073 / Net I/σ(I): 12 |
| Reflection shell | Resolution: 1.93→1.96 Å / Redundancy: 1.5 % / Rmerge(I) obs: 0.324 / Mean I/σ(I) obs: 2.2 / % possible all: 69.1 |
| Reflection shell | *PLUS % possible obs: 69.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 1.95→69 Å / SU B: 7.199 / SU ML: 0.203 / Cross valid method: THROUGHOUT / ESU R Free: 0.191 Details: RESIDUES 356-360 ARE NOT VISIBLE AND ARE NOT MODELLED. NEIGBOURING REGIONS AS WELL AS RESIDUES 384- 390 HAVE POORLY DEFINED STEREOCHEMISTY.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→69 Å
| ||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.197 / Rfactor Rfree: 0.26 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
