 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ghx: A NOVEL SERINE PROTEASE INHIBITION MOTIF INVOLVING A MULTI-CENTER... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ghx | ||||||
|---|---|---|---|---|---|---|---|
| Title | A NOVEL SERINE PROTEASE INHIBITION MOTIF INVOLVING A MULTI-CENTERED SHORT HYDROGEN BONDING NETWORK AT THE ACTIVE SITE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / three-centered / very short hydrogen bond / oxyanion hole water / shift of pKa of His57 /  structure-based drug design / specificity / urokinase / trypsin / thrombin / Zn+2-mediated inhibition / BLOOD CLOTTING / HYDROLASE-HYDROLASE INHIBITOR COMPLEX structure-based drug design / specificity / urokinase / trypsin / thrombin / Zn+2-mediated inhibition / BLOOD CLOTTING / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of lipid kinase activity / positive regulation of phospholipase C-activating G protein-coupled receptor signaling pathway / cytolysis by host of symbiont cells / thrombospondin receptor activity / Defective factor XII causes hereditary angioedema / thrombin / neutrophil-mediated killing of gram-negative bacterium / regulation of blood coagulation / ligand-gated ion channel signaling pathway / Defective F8 cleavage by thrombin ...positive regulation of lipid kinase activity / positive regulation of phospholipase C-activating G protein-coupled receptor signaling pathway / cytolysis by host of symbiont cells / thrombospondin receptor activity / Defective factor XII causes hereditary angioedema / thrombin / neutrophil-mediated killing of gram-negative bacterium / regulation of blood coagulation / ligand-gated ion channel signaling pathway / Defective F8 cleavage by thrombin / Platelet Aggregation (Plug Formation) / negative regulation of platelet activation / negative regulation of astrocyte differentiation / negative regulation of cytokine production involved in inflammatory response / positive regulation of collagen biosynthetic process / positive regulation of blood coagulation / negative regulation of fibrinolysis / Gamma-carboxylation of protein precursors / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / Common Pathway of Fibrin Clot Formation / Removal of aminoterminal propeptides from gamma-carboxylated proteins / regulation of cytosolic calcium ion concentration / fibrinolysis / Intrinsic Pathway of Fibrin Clot Formation / Peptide ligand-binding receptors / positive regulation of release of sequestered calcium ion into cytosol / Regulation of Complement cascade / acute-phase response / Cell surface interactions at the vascular wall / lipopolysaccharide binding / negative regulation of proteolysis / positive regulation of receptor signaling pathway via JAK-STAT / growth factor activity / serine-type endopeptidase inhibitor activity / positive regulation of insulin secretion / platelet activation / response to wounding / Golgi lumen / positive regulation of protein localization to nucleus / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / positive regulation of reactive oxygen species metabolic process / blood coagulation / antimicrobial humoral immune response mediated by antimicrobial peptide / Thrombin signalling through proteinase activated receptors (PARs) / heparin binding / regulation of cell shape / positive regulation of cell growth / G alpha (q) signalling events / collagen-containing extracellular matrix / blood microparticle / cell surface receptor signaling pathway / positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction / positive regulation of protein phosphorylation / G protein-coupled receptor signaling pathway / endoplasmic reticulum lumen / signaling receptor binding / serine-type endopeptidase activity / calcium ion binding / positive regulation of cell population proliferation / proteolysis / extracellular space / extracellular exosome / extracellular region / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Hirudo medicinalis (medicinal leech) Hirudo medicinalis (medicinal leech) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 1.65 Å | ||||||
 Authors Authors | Katz, B.A. / Elrod, K. / Luong, C. / Rice, M. / Mackman, R.L. / Sprengeler, P.A. / Spencer, J. / Hatayte, J. / Janc, J. / Link, J. ...Katz, B.A. / Elrod, K. / Luong, C. / Rice, M. / Mackman, R.L. / Sprengeler, P.A. / Spencer, J. / Hatayte, J. / Janc, J. / Link, J. / Litvak, J. / Rai, R. / Rice, K. / Sideris, S. / Verner, E. / Young, W. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: A novel serine protease inhibition motif involving a multi-centered short hydrogen bonding network at the active site. Authors: Katz, B.A. / Elrod, K. / Luong, C. / Rice, M.J. / Mackman, R.L. / Sprengeler, P.A. / Spencer, J. / Hataye, J. / Janc, J. / Link, J. / Litvak, J. / Rai, R. / Rice, K. / Sideris, S. / Verner, E. / Young, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ghx.cif.gz | 149.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ghx.ent.gz | 119.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1ghx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gh/1ghxftp://data.pdbj.org/pub/pdb/validation_reports/gh/1ghx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ghvC  1ghwC  1ghyC  1ghzC  1gi0C  1gi1C  1gi2C  1gi3C  1gi4C  1gi5C  1gi6C  1gi7C  1gi8C  1gi9C C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein/peptide , 2 types, 2 molecules LI
| #1: Protein/peptide | / E.C.3.4.21.5 / COAGULATION FACTOR II Mass: 4096.534 Da / Num. of mol.: 1 / Fragment: LIGHT CHAIN, RESIDUES 328-363 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P00734, thrombin |
|---|---|
| #3: Protein/peptide | Mass: 1491.528 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) Hirudo medicinalis (medicinal leech) / References: UniProt: P28504 |
-Protein , 1 types, 1 molecules H
| #2: Protein | / E.C.3.4.21.5 / COAGULATION FACTOR II Mass: 29594.055 Da / Num. of mol.: 1 / Fragment: HEAVY CHAIN, RESIDUES 364-620 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P00734, thrombin |
|---|
-Non-polymers , 5 types, 269 molecules 



| #4: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
|---|---|
| #5: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #6: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na |
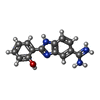
| #7: Chemical | ChemComp-BMZ /  Mass: 253.279 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H13N4O Mass: 253.279 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H13N4O |
| #8: Water | ChemComp-HOH / WaterMass: 18.015 Da / Num. of mol.: 265 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.43 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 7.3 Details: PEG 4000, NaCl (soak at pH 8.2), pH 7.3, vapor diffusion, temperature 298K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / PH range low: 8.2 / PH range high: 7.5 | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Mar 1, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.68→50 Å / Num. all: 87093 / Num. obs: 36570 / % possible obs: 42 % / Observed criterion σ(I): 1 / Redundancy: 2 % / Rmerge(I) obs: 0.067 / Net I/σ(I): 7 |
| Reflection shell | Resolution: 1.57→1.69 Å / Rmerge(I) obs: 0.364 / Num. unique all: 2792 / % possible all: 27.3 |
| Reflection | *PLUS Highest resolution: 1.32 Å / Redundancy: 2 % / Num. measured all: 71823 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.65→7 Å / σ(F): 1.8 / Stereochemistry target values: X-PLOR force field Details: Glu_H18, Asp_H21, Met_H84, and Val_H157 were simultaneously refined in two conformations. Lys_L9 is also disordered. In one conformation it makes an unusual coordinate bond to Zn+2. (Zn+2 ...Details: Glu_H18, Asp_H21, Met_H84, and Val_H157 were simultaneously refined in two conformations. Lys_L9 is also disordered. In one conformation it makes an unusual coordinate bond to Zn+2. (Zn+2 had been added to the crystal in the hope of binding at the active site. The Zn+2 coordinated by Lys_L9 is on the surface and there is no Zn+2 at the active site). (Recently Dan Rich and coworkers found that the amidine of BABIM can coordinate Zn+2) No density was observed for Trp148, Thr149, Ala149A, Asn149B, Val149C, Gly149D, and Lys149E in the autolysis loop, and these residues are not included in the model. No density was observed for C-terminal residues of the heavy chain following Phe_H245. Residues after Phe_H245 are not included in the model. HOH477 makes a short hydrogen bond with OgSer195 and with O6' of the inhibitor Disordered waters include: HOH394 which is in a special position. (It is close to a symmetry related equivalent of itself); HOH395 is close to a symmetry related equivalent of itself; HOH396 is close to a symmetry related equivalent of itself; HOH397 is close to a symmetry related equivalent of itself. The above "waters" correspond to density that is more electron dense than waters. The occupancies were allowed to refine to values greater than unity. HOH674 which is close to HOH675; HOH1127 which is close to a symmetry-related equivalent of HOH1128; HIS_H57 IS doubly protonated. HIS_H91 and His_H119 are MONOPROTONATED ON the epsilon nitrogen
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→7 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 7 Å / σ(F): 1.8 / Rfactor all: 0.211 / Rfactor obs: 0.207 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.65 Å / Lowest resolution: 1.72 Å / Rfactor Rfree: 0.255 / Rfactor obs: 0.212 |
