 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cd0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF HUMAN LAMDA-6 LIGHT CHAIN DIMER JTO | ||||||
 Components Components | PROTEIN (JTO, A VARIABLE DOMAIN FROM LAMBDA-6 TYPE IMMUNOGLOBULIN LIGHT CHAIN) | ||||||
 Keywords Keywords |  IMMUNE SYSTEM / IMMUNOGLOBULIN / BENCE-JONES PROTEIN / LAMDA-6 IMMUNE SYSTEM / IMMUNOGLOBULIN / BENCE-JONES PROTEIN / LAMDA-6 | ||||||
| Function / homology |  Function and homology information Function and homology informationCD22 mediated BCR regulation / Fc epsilon receptor (FCERI) signaling / Classical antibody-mediated complement activation / immunoglobulin complex / Initial triggering of complement / FCGR activation / Role of phospholipids in phagocytosis / Role of LAT2/NTAL/LAB on calcium mobilization / Scavenging of heme from plasma / antigen binding ...CD22 mediated BCR regulation / Fc epsilon receptor (FCERI) signaling / Classical antibody-mediated complement activation / immunoglobulin complex / Initial triggering of complement / FCGR activation / Role of phospholipids in phagocytosis / Role of LAT2/NTAL/LAB on calcium mobilization / Scavenging of heme from plasma / antigen binding / FCERI mediated Ca+2 mobilization / FCGR3A-mediated IL10 synthesis / Antigen activates B Cell Receptor (BCR) leading to generation of second messengers / Regulation of Complement cascade / Cell surface interactions at the vascular wall / FCGR3A-mediated phagocytosis / FCERI mediated MAPK activation / Regulation of actin dynamics for phagocytic cup formation / FCERI mediated NF-kB activation / Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell / Potential therapeutics for SARS / adaptive immune response / immune response / extracellular space / extracellular region / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Pokkuluri, P.R. / Solomon, A. / Weiss, D.T. / Stevens, F.J. / Schiffer, M. | ||||||
 Citation Citation | Journal: Amyloid / Year: 1999 Title: Tertiary structure of human lambda 6 light chains. Authors: Pokkuluri, P.R. / Solomon, A. / Weiss, D.T. / Stevens, F.J. / Schiffer, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cd0.cif.gz | 60.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cd0.ent.gz | 42.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1cd0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cd/1cd0ftp://data.pdbj.org/pub/pdb/validation_reports/cd/1cd0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2cd0C  1dclS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj



















- Assembly
Assembly
| Deposited unit | 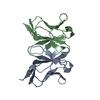
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | BIOLOGICALLY ACTIVE MOLECULE IS A HOMODIMER. ASYMMETRIC UNIT CONTAINS BOTH MONOMERS, CHAINS A AND B. BOTH MONOMERS REFINED INDEPENDENTLY. |
-Components
| #1: Antibody | Mass: 12097.149 Da / Num. of mol.: 2 / Fragment: IMMUNOGLOBULIN VARIABLE DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  Escherichia coli (E. coli) / References: UniProt: P06317, UniProt: P01721*PLUS Escherichia coli (E. coli) / References: UniProt: P06317, UniProt: P01721*PLUS#2: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 247 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 247 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.97 Å3/Da / Density % sol: 38 % Description: CONSTANT DOMAINS WERE ALSO DELETED FROM THE STARTING MODEL. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: 27.5% PEG6000, 0.08 M SODIUM CACODYLATE PH 6.5, 0.1 M SODIUM ACETATE | ||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion / PH range low: 8.5 / PH range high: 4.6 | ||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Oct 1, 1997 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→30 Å / Num. obs: 15655 / % possible obs: 95 % / Redundancy: 4.87 % / Biso Wilson estimate: 8.4 Å2 / Rmerge(I) obs: 0.076 / Net I/σ(I): 13.5 |
| Reflection shell | Resolution: 1.9→1.94 Å / Redundancy: 3 % / Rmerge(I) obs: 0.311 / Mean I/σ(I) obs: 3.3 / % possible all: 89.8 |
| Reflection shell | *PLUS % possible obs: 89.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: MCG(PDBCODE:1DCL): VL DIMER WITH WITH CDR LOOPS DELETED. Resolution: 1.9→8 Å / Rfactor Rfree error: 0.007 / Data cutoff high rms absF: 362162.81 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 3 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 72.6 Å2 / ksol: 0.46 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.93 Å / Rfactor Rfree error: 0.044 / Total num. of bins used: 24
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
