 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a25 | ||||||
|---|---|---|---|---|---|---|---|
| Title | C2 DOMAIN FROM PROTEIN KINASE C (BETA) | ||||||
 Components Components | PROTEIN KINASE C (BETA) | ||||||
 Keywords Keywords |  CALCIUM-BINDING PROTEIN / CALCIUM++/PHOSPHOLIPID BINDING PROTEIN CALCIUM-BINDING PROTEIN / CALCIUM++/PHOSPHOLIPID BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationdibenzo-p-dioxin metabolic process / Depolymerization of the Nuclear Lamina / Disinhibition of SNARE formation / Response to elevated platelet cytosolic Ca2+ / histone H3T6 kinase activity / Activation of NF-kappaB in B cells / VEGFR2 mediated cell proliferation / positive regulation of odontogenesis of dentin-containing tooth / positive regulation of B cell receptor signaling pathway / spectrin ...dibenzo-p-dioxin metabolic process / Depolymerization of the Nuclear Lamina / Disinhibition of SNARE formation / Response to elevated platelet cytosolic Ca2+ / histone H3T6 kinase activity / Activation of NF-kappaB in B cells / VEGFR2 mediated cell proliferation / positive regulation of odontogenesis of dentin-containing tooth / positive regulation of B cell receptor signaling pathway / spectrin / regulation of glucose transmembrane transport / WNT5A-dependent internalization of FZD4 / RHO GTPases Activate NADPH Oxidases / protein kinase C / phospholipase C-activating G protein-coupled acetylcholine receptor signaling pathway / negative regulation of glucose transmembrane transport / cellular response to carbohydrate stimulus / diacylglycerol-dependent serine/threonine kinase activity / response to vitamin D / presynaptic cytosol / positive regulation of vascular endothelial growth factor receptor signaling pathway / regulation of growth / nuclear androgen receptor binding / regulation of synaptic vesicle exocytosis / regulation of dopamine secretion / B cell activation / Trafficking of GluR2-containing AMPA receptors / calcium channel regulator activity / calyx of Held / response to glucose / negative regulation of insulin receptor signaling pathway / presynaptic modulation of chemical synaptic transmission / post-translational protein modification / protein kinase C binding / nuclear receptor coactivator activity / brush border membrane / B cell receptor signaling pathway / intracellular calcium ion homeostasis / positive regulation of insulin secretion / positive regulation of angiogenesis / calcium ion transport / histone binding / positive regulation of canonical NF-kappaB signal transduction / response to ethanol / adaptive immune response / intracellular signal transduction / response to xenobiotic stimulus / phosphorylation / protein serine kinase activity / centrosome / protein serine/threonine kinase activity / apoptotic process / chromatin binding / regulation of transcription by RNA polymerase II / zinc ion binding / nucleoplasm / ATP binding / membrane / nucleus / plasma membrane / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Rattus norvegicus (Norway rat) Rattus norvegicus (Norway rat) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Sutton, R.B. / Sprang, S.R. | ||||||
 Citation Citation | Journal: Structure / Year: 1998 Title: Structure of the protein kinase Cbeta phospholipid-binding C2 domain complexed with Ca2+. Authors: Sutton, R.B. / Sprang, S.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a25.cif.gz | 67.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a25.ent.gz | 51.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1a25.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a2/1a25ftp://data.pdbj.org/pub/pdb/validation_reports/a2/1a25 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1rsyS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.912012, 0.409902, -0.014639), Vector : |
-Components
| #1: Protein | Mass: 16882.111 Da / Num. of mol.: 2 / Fragment: CALCIUM/PHOSPHOLIPID BINDING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rattus norvegicus (Norway rat) / Cell line: BL21 / Plasmid: PGEX-KG / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3) Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21 (DE3)References: UniProt: P68403, Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor#2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-PSE / | 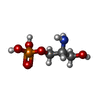  Mass: 171.089 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H10NO5P Mass: 171.089 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H10NO5P#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.5 Å3/Da / Density % sol: 64 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: PROTEIN WAS CRYSTALLIZED FROM 15% PEG1500, 100 MM MES, PH 6.5 | ||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 21 ℃ / pH: 6.4 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 133 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 0.918 / Beamline: F1 / Wavelength: 0.918 |
| Detector | Type: PRINCETON 2K / Detector: CCD / Date: Oct 1, 1996 / Details: MIRRORS |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.918 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→40 Å / Num. obs: 12297 / % possible obs: 98.5 % / Observed criterion σ(I): 2 / Redundancy: 8.9 % / Biso Wilson estimate: 50.7 Å2 / Rmerge(I) obs: 0.136 / Rsym value: 0.083 / Net I/σ(I): 13 |
| Reflection shell | Resolution: 2.7→2.9 Å / Redundancy: 5 % / Rmerge(I) obs: 0.13 / Mean I/σ(I) obs: 8 / Rsym value: 0.26 / % possible all: 95 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1RSY Resolution: 2.7→20 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1365552.04 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: REFINEMENT TARGET FUNCTION : MLF DATA CUTOFF HIGH (ABS(F)) : 1365552.04 DATA CUTOFF LOW (ABS(F)) : 0.000000
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 25 Å2 / ksol: 0.345 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: CONSTRAINED | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.87 Å / Rfactor Rfree error: 0.031 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.2 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
