 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1a0j: CRYSTAL STRUCTURE OF A NON-PSYCHROPHILIC TRYPSIN FROM A COLD-ADAP... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a0j | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A NON-PSYCHROPHILIC TRYPSIN FROM A COLD-ADAPTED FISH SPECIES. | ||||||
 Components Components | TRYPSIN | ||||||
 Keywords Keywords | SERINE PROTEASE / SERINE PROTEINASE / TRYPSIN / HYDROLASE | ||||||
| Function / homology |  Function and homology informationtrypsin / digestion / serine-type endopeptidase activity / extracellular space / metal ion binding Function and homology informationtrypsin / digestion / serine-type endopeptidase activity / extracellular space / metal ion bindingSimilarity search - Function | ||||||
| Biological species |  Salmo salar (Atlantic salmon) Salmo salar (Atlantic salmon) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Schroeder, H.-K. / Willassen, N.P. / Smalaas, A.O. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 1998 Title: Structure of a non-psychrophilic trypsin from a cold-adapted fish species. Authors: Schroder, H.K. / Willassen, N.P. / Smalas, A.O. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a0j.cif.gz | 183.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a0j.ent.gz | 148.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1a0j.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a0/1a0jftp://data.pdbj.org/pub/pdb/validation_reports/a0/1a0j | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 23803.662 Da / Num. of mol.: 4 / Source method: isolated from a natural source Details: BENZAMIDINE INHIBITOR IN THE ACTIVE SITE. THE AMINO ACID NUMBERING SCHEME USED IS ADOPTED FROM CHYMOTRYPSINOGEN. Source: (natural) Salmo salar (Atlantic salmon) / Organ: PANCREAS / References: UniProt: P35033, trypsin#2: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-SO4 / Sulfate  Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-BEN / Benzamidine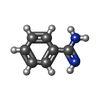  Mass: 120.152 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C7H8N2#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 379 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 379 / Source method: isolated from a natural source / Formula: H2OSequence details | THE AMINO ACID NUMBERING SCHEME USED IS ADOPTED FROM CHYMOTRYPS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 4 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.6 / Details: pH 4.6 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 310 K / Method: vapor diffusionDetails: drop contained 1:1 mixture of protein and reservoir solution | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM1A / Wavelength: 0.873 / Beamline: BM1A / Wavelength: 0.873 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 1, 1996 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.873 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→28 Å / Num. obs: 76357 / % possible obs: 81.2 % / Observed criterion σ(I): 1 / Redundancy: 5.9 % / Biso Wilson estimate: 19.6 Å2 / Rmerge(I) obs: 0.123 / Net I/σ(I): 3.6 |
| Reflection shell | Resolution: 1.7→1.79 Å / Redundancy: 2 % / Rmerge(I) obs: 0.258 / Mean I/σ(I) obs: 2.9 / % possible all: 52.3 |
| Reflection | *PLUS Num. measured all: 451079 |
| Reflection shell | *PLUS % possible obs: 52.3 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3PTB BOVINE TRYPSIN, WITH CORRECT AMINO ACID SEQUENCE SUPERIMPOSED ONTO ANIONIC SALMON TRYPSIN 2TBS. Resolution: 1.7→8 Å / Cross valid method: THROUGHOUT / σ(F): 3 Details: NCS RESTRAINTS WERE INCLUDED IN THE FIRST REFINEMENT ROUNDS, THEN LEFT OUT. THE ELECTRON DENSITY INDICATES A CALCIUM ION FOR ONLY ONE OF THE FOUR MOLECULES (MOL A). THE ELECTRON DENSITY ...Details: NCS RESTRAINTS WERE INCLUDED IN THE FIRST REFINEMENT ROUNDS, THEN LEFT OUT. THE ELECTRON DENSITY INDICATES A CALCIUM ION FOR ONLY ONE OF THE FOUR MOLECULES (MOL A). THE ELECTRON DENSITY INDICATES A CALCIUM ION FOR ONLY ONE OF THE FOUR MOLECULES (MOL A).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.93 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.18 Å / Luzzati sigma a free: 0.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.76 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.8 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.2347 |
