 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-5301 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
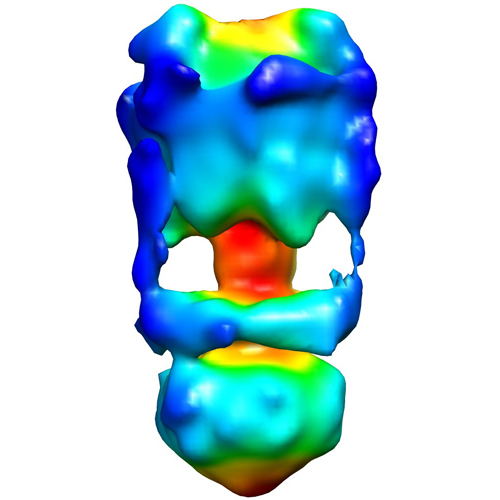
| タイトル | Negative Stain reconstruction of the Thermus thermophilus A-ATPase to 23 Angstrom. Opposite Hand to published. | |||||||||
 マップデータ マップデータ | This is a negative stain reconstruction of the Thermus thermophilus A-ATPase | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード |  A-ATPase / Thermus thermophilus (サーマス・サーモフィルス) / ATPase (ATPアーゼ) A-ATPase / Thermus thermophilus (サーマス・サーモフィルス) / ATPase (ATPアーゼ) | |||||||||
| 生物種 |   Thermus thermophilus (サーマス・サーモフィルス) Thermus thermophilus (サーマス・サーモフィルス) | |||||||||
| 手法 | 単粒子再構成法 / ネガティブ染色法 / 解像度: 23.0 Å | |||||||||
 データ登録者 データ登録者 | Bernal RA / Stock D | |||||||||
 引用 引用 | ジャーナル: Structure / 年: 2004 タイトル: Three-dimensional structure of the intact Thermus thermophilus H+-ATPase/synthase by electron microscopy. 著者: Ricardo A Bernal / Daniela Stock /  要旨: ATPases are unique rotary motors that are essential to all living organisms because of their role in energy interconversion. A three-dimensional reconstruction of the intact H+-ATPase/synthase from ...ATPases are unique rotary motors that are essential to all living organisms because of their role in energy interconversion. A three-dimensional reconstruction of the intact H+-ATPase/synthase from Thermus thermophilus has revealed the presence of two interconnected peripheral stalks, a well-defined central stalk, and a hexagonally shaped hydrophobic domain. The peripheral stalks are each attached to the water soluble sector at a noncatalytic subunit interface and extend down toward the membrane where they interact with a strong elongated tube of density that runs parallel to the membrane and connects the two stalks. The central stalk is well resolved, especially with respect to its interaction with a single catalytic subunit giving rise to an asymmetry comparable to that identified in F-ATPases. The hexagonal shape of the membrane domain might suggest the presence of 12 proteolipids arranged as dimers, analogous to the proposed arrangement in the related eukaryotic V-ATPases. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_5301.map.gz | 7.5 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-5301-v30.xmlemd-5301.xml | 14.3 KB 14.3 KB | 表示 表示 | EMDBヘッダ |
| 画像 | 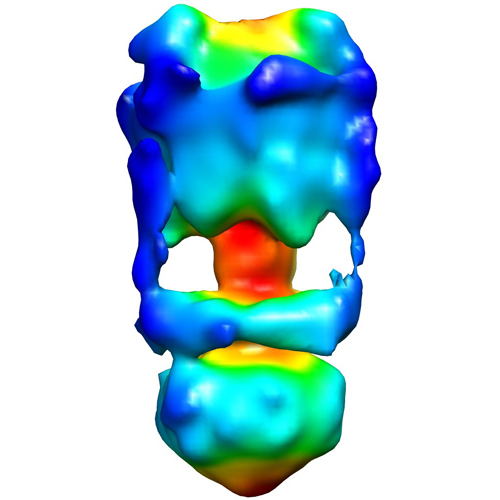 emd_5301_1.jpg emd_5301_1.jpg | 118.2 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-5301ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5301 http://ftp.pdbj.org/pub/emdb/structures/EMD-5301ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5301 | HTTPS FTP |
-関連構造データ







-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_5301.map.gz / 形式: CCP4 / 大きさ: 7.8 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | This is a negative stain reconstruction of the Thermus thermophilus A-ATPase | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 3.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Thermus thermophilus A-ATPase
| 全体 | 名称: Thermus thermophilus A-ATPase |
|---|---|
| 要素 |
|
-超分子 #1000: Thermus thermophilus A-ATPase
| 超分子 | 名称: Thermus thermophilus A-ATPase / タイプ: sample / ID: 1000 / 集合状態: multi-subunit complex / Number unique components: 1 |
|---|
-超分子 #1: ATPase
| 超分子 | 名称: ATPase / タイプ: organelle_or_cellular_component / ID: 1 / Name.synonym: ATPase / 集合状態: multimer / 組換発現: No / データベース: NCBI |
|---|---|
| 由来(天然) | 生物種: Thermus thermophilus (サーマス・サーモフィルス) 株: HB8 / 別称: Thermus thermophilus / 細胞中の位置: membrane |
-実験情報
-構造解析
| 手法 | ネガティブ染色法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.02 mg/mL |
|---|---|
| 緩衝液 | pH: 8 詳細: 20 mM Tris, pH 8.0, 2 mM MgCl2, 1 mM EDTA, 0.05% n-dodecyl-beta-D-maltoside, 0.02% NaN3 |
| 染色 | タイプ: NEGATIVE 詳細: Three microliters of the T. thermophilus sample, diluted to 0.02 mg/mL, was placed onto the surface of the carbon-coated grid. The sample was blotted off and replaced with 3 microliters of 2% ...詳細: Three microliters of the T. thermophilus sample, diluted to 0.02 mg/mL, was placed onto the surface of the carbon-coated grid. The sample was blotted off and replaced with 3 microliters of 2% uranyl acetate. The uranyl acetate was blotted away and replaced with 3 microliters of 4% methylamine tungstate. The final drop of methylamine tungstate was blotted away and the grid was left to air dry. |
| グリッド | 詳細: 400 mesh 3.05 mm copper grids with a thin layer carbon support |
| 凍結 | 凍結剤: NONE / 装置: OTHER |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI 12 |
|---|---|
| 電子線 | 加速電圧: 120 kV / 電子線源: LAB6 |
| 電子光学系 | 倍率(補正後): 42000 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELDBright-field microscopy / 最大 デフォーカス(公称値): 1.0 µm / 最小 デフォーカス(公称値): 1.0 µm / 倍率(公称値): 42000 |
| 試料ステージ | 試料ホルダー: side entry single tilt / 試料ホルダーモデル: OTHER |
| 温度 | 平均: 25 K |
| アライメント法 | Legacy - 非点収差: not corrected |
| 日付 | 2003年1月22日 |
| 撮影 | カテゴリ: FILM / フィルム・検出器のモデル: KODAK SO-163 FILM / デジタル化 - スキャナー: ZEISS SCAI / デジタル化 - サンプリング間隔: 14 µm / ビット/ピクセル: 8 |
-画像解析
| CTF補正 | 詳細: no ctf correction done because it was a negative stain reconstruction |
|---|---|
| 最終 2次元分類 | クラス数: 60 |
| 最終 再構成 | アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 23.0 Å / 解像度の算出法: FSC 0.5 CUT-OFF / ソフトウェア - 名称: MRC Image2000 and Imagic / 使用した粒子像数: 12300 |
-原子モデル構築 1
| 初期モデル | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C / Chain - #3 - Chain ID: D / Chain - #4 - Chain ID: E / Chain - #5 - Chain ID: F / Chain - #6 - Chain ID: G |
|---|---|
| ソフトウェア | 名称: EMfit |
| 詳細 | PDBEntryID_givenInChain. Protocol: Each chain was fit as a separate rigid body. X-ray coordinates for the bovine alpha3 beta3 and gamma sub-assemblies were manually fitted into the EM density using the program O. The program EMfit (Rossmann et al., 2001) was then used in order to obtain a more quantitative fit. |
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT 当てはまり具合の基準: sumf and number of atoms inside density |

-原子モデル構築 2
| 初期モデル | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C |
|---|---|
| ソフトウェア | 名称: EMfit |
| 詳細 | PDBEntryID_givenInChain. Protocol: Each chain was fit as a separate rigid body. X-ray coordinates for the bovine alpha3 beta3 and gamma sub-assemblies were manually fitted into the EM density using the program O. The program EMfit (Rossmann et al., 2001) was then used in order to obtain a more quantitative fit. |
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT 当てはまり具合の基準: sumf and number of atoms inside density |

-原子モデル構築 3
| 初期モデル | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C / Chain - #3 - Chain ID: D / Chain - #4 - Chain ID: E / Chain - #5 - Chain ID: F / Chain - #6 - Chain ID: G / Chain - #7 - Chain ID: H / Chain - #8 - Chain ID: I / Chain - #9 - Chain ID: J / Chain - #10 - Chain ID: K / Chain - #11 - Chain ID: L |
|---|---|
| ソフトウェア | 名称: EMfit |
| 詳細 | PDBEntryID_givenInChain. Protocol: Each chain was fit as a separate rigid body. X-ray coordinates for the bovine alpha3 beta3 and gamma sub-assemblies were manually fitted into the EM density using the program O. The program EMfit (Rossmann et al., 2001) was then used in order to obtain a more quantitative fit. |
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT 当てはまり具合の基準: sumf and number of atoms inside density |

