 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4qg7: S.aureus TMK in complex with a potent inhibitor compound 18, 2-(3... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4qg7 | ||||||
|---|---|---|---|---|---|---|---|
| Title | S.aureus TMK in complex with a potent inhibitor compound 18, 2-(3-CHLOROPHENOXY)-3-METHOXY-4-{[(3S)-3-(5-METHYL-2,4-DIOXO-3,4-DIHYDROPYRIMIDIN-1(2H)-YL)PIPERIDIN-1-YL]METHYL}BENZOIC ACID | ||||||
 Components Components | Thymidylate kinase | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / TRANSFERASE/TRANSFERASE INHIBITOR kinase / thymidine monphosphate / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationdUDP biosynthetic process / dTMP kinase / thymidylate kinase activity / dTDP biosynthetic process / dTTP biosynthetic process / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus subsp. aureus (bacteria) Staphylococcus aureus subsp. aureus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.67 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.67 Å | ||||||
 Authors Authors | Olivier, N.B. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2014 Title: Antibacterial inhibitors of gram-positive thymidylate kinase: structure-activity relationships and chiral preference of a new hydrophobic binding region. Authors: Kawatkar, S.P. / Keating, T.A. / Olivier, N.B. / Breen, J.N. / Green, O.M. / Guler, S.Y. / Hentemann, M.F. / Loch, J.T. / McKenzie, A.R. / Newman, J.V. / Otterson, L.G. / Martinez-Botella, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4qg7.cif.gz | 96.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4qg7.ent.gz | 73.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4qg7.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qg/4qg7ftp://data.pdbj.org/pub/pdb/validation_reports/qg/4qg7 | HTTPS FTP |
|---|
-Related structure data














-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23454.586 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus subsp. aureus (bacteria)Strain: MRSA252 / Gene: SAR0483, tmk / Plasmid: pET28a / Production host: #2: Chemical | 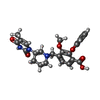  Mass: 499.943 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H26ClN3O6 Mass: 499.943 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C25H26ClN3O6#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.1 Å3/Da / Density % sol: 41.41 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: To obtain the inhibitor bound crystal form of TMK-S.aureus crystals were initially grown in the absence of compound using the sitting drop method at 293 K with a reservoir solution of 100 mM ...Details: To obtain the inhibitor bound crystal form of TMK-S.aureus crystals were initially grown in the absence of compound using the sitting drop method at 293 K with a reservoir solution of 100 mM PCPT (propionate-cacodylate-bistris propane buffer) pH 7-8, 21-24% PEG 3350, 200 mM Mg2Cl using 1:1 protein:reservoir solution with the protein solution at 13 mg/mL. Crystals were harvested and soaked overnight in a solution containing 100 mM PCPT, 35% PEG 3350, 200 mM Mg2Cl and 1-2 mM or compound from a 100 mM DMSO stock. After soaking the crystals were cryoprotected by soaking for 15 minutes in compound-soak solution supplemented with 20% ethylene glycol., VAPOR DIFFUSION, SITTING DROP PH range: 7-8 |
-Data collection
| Diffraction | Mean temperature: 140 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E+ SUPERBRIGHT / Wavelength: 1.54 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU SATURN 944+ / Detector: CCD / Date: Aug 17, 2010 / Details: monochrome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: mirror / Protocol: SINGLE WAVELENGTH / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.67→90.45 Å / Num. all: 40350 / Num. obs: 40350 / % possible obs: 89.8 % / Redundancy: 2.8 % / Biso Wilson estimate: 21.7 Å2 / Rsym value: 0.04 / Net I/σ(I): 14.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: in-house apostate Resolution: 1.67→45.22 Å / Cor.coef. Fo:Fc: 0.9246 / Cor.coef. Fo:Fc free: 0.9155 / SU R Cruickshank DPI: 0.116 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 137.63 Å2 / Biso mean: 27.08 Å2 / Biso min: 10.32 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.205 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.67→45.22 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.67→1.71 Å / Total num. of bins used: 20
|
