 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1994: Negative stain reconstruction of the Saccharomyces cerevisiae pro... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1994 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
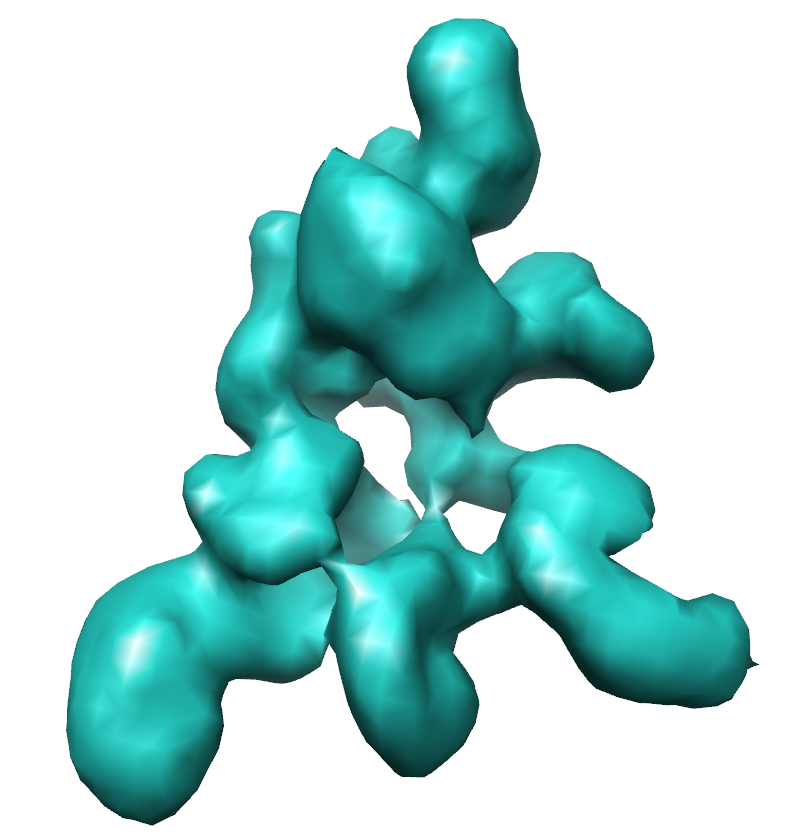
| Title | Negative stain reconstruction of the Saccharomyces cerevisiae proteasome lid | |||||||||
 Map data Map data | map of endogenous Saccharomyces cerevisiae proteasome lid | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | 26S / 19S /  proteasome / yeast lid / regulatory particle / ubiquitin recognition / deubiquitination / AAA-ATPase proteasome / yeast lid / regulatory particle / ubiquitin recognition / deubiquitination / AAA-ATPase | |||||||||
| Function / homology | Proteasome/cyclosome repeat / proteasome regulatory particle, lid subcomplex Function and homology information Function and homology information | |||||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 15.0 Å | |||||||||
 Authors Authors | Lander GC / Estrin E / Matyskiela M / Bashore C / Nogales E / Martin A | |||||||||
 Citation Citation | Journal: Nature / Year: 2012 Title: Complete subunit architecture of the proteasome regulatory particle. Authors: Gabriel C Lander / Eric Estrin / Mary E Matyskiela / Charlene Bashore / Eva Nogales / Andreas Martin /  Abstract: The proteasome is the major ATP-dependent protease in eukaryotic cells, but limited structural information restricts a mechanistic understanding of its activities. The proteasome regulatory particle, ...The proteasome is the major ATP-dependent protease in eukaryotic cells, but limited structural information restricts a mechanistic understanding of its activities. The proteasome regulatory particle, consisting of the lid and base subcomplexes, recognizes and processes polyubiquitinated substrates. Here we used electron microscopy and a new heterologous expression system for the lid to delineate the complete subunit architecture of the regulatory particle from yeast. Our studies reveal the spatial arrangement of ubiquitin receptors, deubiquitinating enzymes and the protein unfolding machinery at subnanometre resolution, outlining the substrate's path to degradation. Unexpectedly, the ATPase subunits within the base unfoldase are arranged in a spiral staircase, providing insight into potential mechanisms for substrate translocation through the central pore. Large conformational rearrangements of the lid upon holoenzyme formation suggest allosteric regulation of deubiquitination. We provide a structural basis for the ability of the proteasome to degrade a diverse set of substrates and thus regulate vital cellular processes. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1994.map.gz | 421.4 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1994-v30.xmlemd-1994.xml | 9.8 KB 9.8 KB | Display Display | EMDB header |
| Images | 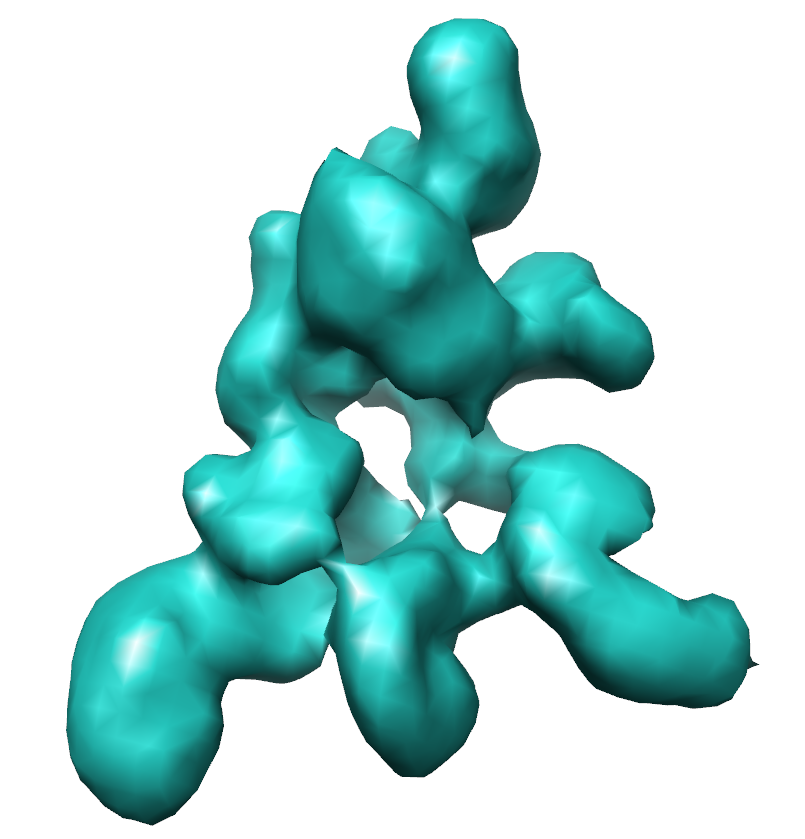 em1994.png em1994.png | 218.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1994ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1994 http://ftp.pdbj.org/pub/emdb/structures/EMD-1994ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1994 | HTTPS FTP |
-Related structure data









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1994.map.gz / Format: CCP4 / Size: 7.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | map of endogenous Saccharomyces cerevisiae proteasome lid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.76 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Saccharomyces cerevisiae proteasome lid subcomplex
| Entire | Name: Saccharomyces cerevisiae proteasome lid subcomplex |
|---|---|
| Components |
|
-Supramolecule #1000: Saccharomyces cerevisiae proteasome lid subcomplex
| Supramolecule | Name: Saccharomyces cerevisiae proteasome lid subcomplex / type: sample / ID: 1000 / Details: monodisperse / Oligomeric state: 8 subunit complex / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 361 KDa / Theoretical: 361 KDa / Method: Mass Spectrometry |
-Macromolecule #1: lid
| Macromolecule | Name: lid / type: protein_or_peptide / ID: 1 / Name.synonym: lid Details: lid was purified from endogenous proteasome holoenzyme using a 1M salt wash Number of copies: 1 / Oligomeric state: monomer / Recombinant expression: No |
|---|---|
| Source (natural) | Organism: Saccharomyces cerevisiae (brewer's yeast) / Strain: YYS40 / synonym: Baker's Yeast |
| Molecular weight | Experimental: 361 KDa / Theoretical: 361 KDa |
| Sequence | GO: proteasome regulatory particle, lid subcomplex / InterPro: Proteasome/cyclosome repeat |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.009 mg/mL |
|---|---|
| Buffer | pH: 7.6 Details: 60mM HEPES, 50mM NaCl, 50mM KCl, 5 mM MgCl2, 0.5mM EDTA, 1mM DTT, 10% glycerol |
| Staining | Type: NEGATIVE Details: Protein adsorbed to grid for 1 minute, then passed over four 50uL drops of 2% w/v uranyl formate, 5 seconds on each drop |
| Grid | Details: 200 mesh Cu grid |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI 20 |
|---|---|
| Electron beam | Acceleration voltage: 120 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 80000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.2 mm / Nominal defocus max: 1.2 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 80000 |
| Sample stage | Specimen holder: Room temp single tilt / Specimen holder model: SIDE ENTRY, EUCENTRIC |
| Temperature | Min: 78 K / Max: 78 K / Average: 78 K |
| Alignment procedure | Legacy - Astigmatism: objective lens astigmatism was corrected at 210,000 times magnification Legacy - Electron beam tilt params: 0 |
| Details | Data acquired using Leginon |
| Date | Apr 2, 2011 |
| Image recording | Category: CCD / Film or detector model: GENERIC GATAN (4k x 4k) / Number real images: 250 / Average electron dose: 20 e/Å2 |
-Image processing
| CTF correction | Details: whole micrograph |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 15.0 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: EMAN2 SPARX / Number images used: 33478 |
| Details | Image processing performed in the Appion processing environment. 3D reconstruction performed using EMAN2 and SPARX libraries |
