 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-8731: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstro... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8731 | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
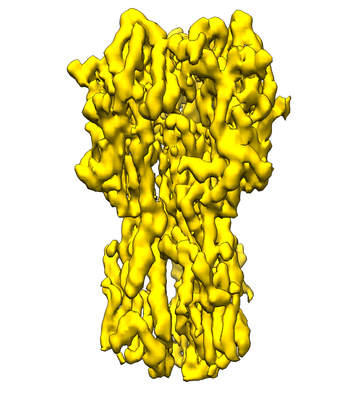
| Title | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees | |||||||||||||||||||||||||||
 Map data Map data | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, sharpened using a B-factor of 320 | |||||||||||||||||||||||||||
 Sample Sample |
| |||||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology information viral budding from plasma membrane / clathrin-dependent endocytosis of virus by host cell / host cell surface receptor binding / apical plasma membrane / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion attachment to host cell / host cell plasma membrane / virion membrane viral budding from plasma membrane / clathrin-dependent endocytosis of virus by host cell / host cell surface receptor binding / apical plasma membrane / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion attachment to host cell / host cell plasma membrane / virion membraneSimilarity search - Function | |||||||||||||||||||||||||||
| Biological species |  Influenza A virus (A/Hong Kong/1/1968(H3N2)) Influenza A virus (A/Hong Kong/1/1968(H3N2)) | |||||||||||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.2 Å | |||||||||||||||||||||||||||
 Authors Authors | Tan Y / Baldwin PR / Potter CS / Carragher B / Lyumkis D | |||||||||||||||||||||||||||
| Funding support |  Singapore, Singapore,  United States, 8 items United States, 8 items
| |||||||||||||||||||||||||||
 Citation Citation | Journal: Nat Methods / Year: 2017 Title: Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Authors: Yong Zi Tan / Philip R Baldwin / Joseph H Davis / James R Williamson / Clinton S Potter / Bridget Carragher / Dmitry Lyumkis / Abstract: We present a strategy for tackling preferred specimen orientation in single-particle cryogenic electron microscopy by employing tilts during data collection. We also describe a tool to quantify the ...We present a strategy for tackling preferred specimen orientation in single-particle cryogenic electron microscopy by employing tilts during data collection. We also describe a tool to quantify the resulting directional resolution using 3D Fourier shell correlation volumes. We applied these methods to determine the structures at near-atomic resolution of the influenza hemagglutinin trimer, which adopts a highly preferred specimen orientation, and of ribosomal biogenesis intermediates, which adopt moderately preferred orientations. | |||||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8731.map.gz | 59.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8731-v30.xmlemd-8731.xml | 33.1 KB 33.1 KB | Display Display | EMDB header |
| Images |  emd_8731.png emd_8731.png | 107.3 KB | ||
| Masks | emd_8731_msk_1.map | 64 MB | Mask map | |
| Others | emd_8731_additional_1.map.gzemd_8731_additional_2.map.gzemd_8731_additional_3.map.gzemd_8731_additional_4.map.gzemd_8731_additional_5.map.gzemd_8731_half_map_1.map.gzemd_8731_half_map_2.map.gz | 49.5 MB 525 KB 991.1 KB 4.1 MB 4.1 MB 49.6 MB 49.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8731ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8731 http://ftp.pdbj.org/pub/emdb/structures/EMD-8731ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8731 | HTTPS FTP |
-Related structure data
| Similar structure data | |
|---|---|
| EM raw data | EMPIAR-10096 (Title: Untilted Single-Particle CryoEM of Highly Preferred Orientated Influenza Hemagglutinin Trimer Data size: 1.2 TB Data #1: Unaligned raw multi-frame untilted micrographs of Influenza Hemagglutinin Trimer [micrographs - multiframe] Data #2: Aligned, summed and exposure weighted untilted micrographs of Influenza Hemagglutinin Trimer [micrographs - single frame] Data #3: Equalized particle stack of Influenza Hemagglutinin Trimer [picked particles - multiframe - processed]) EMPIAR-10097 (Title: 40 Degree Tilted Single-Particle CryoEM of Highly Preferred Orientated Influenza Hemagglutinin TrimerData size: 1.8 TB Data #1: Unaligned raw multi-frame 40 degree tilted micrographs of Influenza Hemagglutinin Trimer [micrographs - multiframe] Data #2: Aligned, summed and exposure weighted 40 degree tilted micrographs of Influenza Hemagglutinin Trimer [micrographs - single frame] Data #3: Equalized particle stack of Influenza Hemagglutinin Trimer from 40 degree tilted micrographs [picked particles - multiframe - processed]) |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |






-Map
| File | Download / File: emd_8731.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, sharpened using a B-factor of 320 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.31 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
-Mask #1
| File | emd_8731_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
 Z
Z Y
Y X
X









-Additional map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, unsharpened | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, map-to-model 3DFSC, thresholded at 0.5 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, half-map 3DFSC, thresholded at 0.143 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






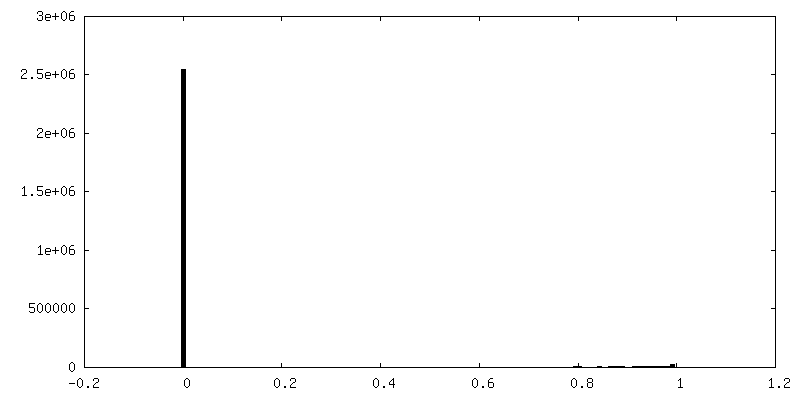

-Additional map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_additional_4.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, map-to-model 3DFSC, raw | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Additional map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_additional_5.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, half-map 3DFSC, raw | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom...
| File | emd_8731_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Influenza hemagglutinin (HA) trimer reconstruction at 4.2 Angstrom resolution using particles from micrographs tilted at 40 degrees, half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : Influenza hemagglutinin (HA) trimer
| Entire | Name: Influenza hemagglutinin (HA) trimer |
|---|---|
| Components |
|
-Supramolecule #1: Influenza hemagglutinin (HA) trimer
| Supramolecule | Name: Influenza hemagglutinin (HA) trimer / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: all Details: A/Hong Kong/1/1968 (H3N2), obtained commercially from MyBioSource, catalog MBS434205 |
|---|---|
| Source (natural) | Organism: Influenza A virus (A/Hong Kong/1/1968(H3N2)) |
| Molecular weight | Theoretical: 150 KDa |
| Recombinant expression | Organism:  Homo sapiens (human) / Recombinant strain: HEK293 Homo sapiens (human) / Recombinant strain: HEK293 |
-Macromolecule #1: Influenza hemagglutinin (HA) Trimer A/Hong Kong/1/1968 H3N2
| Macromolecule | Name: Influenza hemagglutinin (HA) Trimer A/Hong Kong/1/1968 H3N2 type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Influenza A virus (A/Hong Kong/1/1968(H3N2)) |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: QDLPGNDNST ATLCLGHHAV PNGTLVKTIT DDQIEVTNAT ELVQSSSTGK ICNNPHRILD GIDCTLIDAL LGDPHCDVFQ NETWDLFVER SKAFSNCYPY DVPDYASLRS LVASSGTLEF ITEGFTWTGV TQNGGSNACK RGPGSGFFSR LNWLTKSGST YPVLNVTMPN ...String: QDLPGNDNST ATLCLGHHAV PNGTLVKTIT DDQIEVTNAT ELVQSSSTGK ICNNPHRILD GIDCTLIDAL LGDPHCDVFQ NETWDLFVER SKAFSNCYPY DVPDYASLRS LVASSGTLEF ITEGFTWTGV TQNGGSNACK RGPGSGFFSR LNWLTKSGST YPVLNVTMPN NDNFDKLYIW GVHHPSTNQE QTSLYVQASG RVTVSTRRSQ QTIIPNIGSR PWVRGLSSRI SIYWTIVKPG DVLVINSNGN LIAPRGYFKM RTGKSSIMRS DAPIDTCISE CITPNGSIPN DKPFQNVNKI TYGACPKYVK QNTLKLATGM RNVPEKQTRG LFGAIAGFIE NGWEGMIDGW YGFRHQNSEG TGQAADLKST QAAIDQINGK LNRVIEKTNE KFHQIEKEFS EVEGRIQDLE KYVEDTKIDL WSYNAELLVA LENQHTIDLT DSEMNKLFEK TRRQLRENAE DMGNGCFKIY HKCDNACIES IRNGTYDHDV YRDEALNNRF QIKGVELKSG YKD |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.75 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
Details: PBS Buffer | |||||||||||||||
| Grid | Model: C-Flat CF-1.2/1.3-4Au / Material: GOLD / Mesh: 400 / Support film - topology: HOLEY / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 80 % / Chamber temperature: 298 K / Instrument: GATAN CRYOPLUNGE 3 Details: 3 microliters of 0.75 mg/mL sample was added to a plasma-cleaned (Gatan Solarus) 1.2-micron-hole, 1.3-micron-spacing holey gold grid (made in-house) and plunge-frozen in liquid ethane using ...Details: 3 microliters of 0.75 mg/mL sample was added to a plasma-cleaned (Gatan Solarus) 1.2-micron-hole, 1.3-micron-spacing holey gold grid (made in-house) and plunge-frozen in liquid ethane using the Cryoplunge 3 system (Gatan) operating at 80% humidity, 298K ambient temperature.. | |||||||||||||||
| Details | Fraction was obtained directly from a sucrose gradient, and was spin-concentrated in a 100 kDa MW-cutoff filter. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated magnification: 38167 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 3.5 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 22500 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Details | In order to account for highly preferred orientations of the specimen, data were acquired using tilts of 40 degrees. The CTF was estimated on a per-particle basis to account for the gradient of CTF values across individual micrographs. |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-100 / Number grids imaged: 2 / Number real images: 847 / Average exposure time: 20.0 sec. / Average electron dose: 82.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 287820 Details: 287820 particles were picked using Gautomatch with templates made from an initial 2D classification of a subset of the particles. These particles were put through 2D classification to produce 158432 particles. |
|---|---|
| CTF correction | Software - Name: Gctf (ver. 0.5) |
| Startup model | Type of model: INSILICO MODEL In silico model: Ab initio model was produced using CryoSPARC using the 158432 particles. |
| Initial angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 2) |
| Final angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 2) |
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: C3 (3 fold cyclic) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 4.2 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 2) Details: Directional anisotropy is present in the resolution, with a range from 3.7 to 5.5 Angstrom, 3D FSC sphericity of 0.97, and map-to-model resolution of 4.4 Angstrom. Number images used: 158432 |
