 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8252 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 2.6A 3D reconstruction of Tulane virus | |||||||||
 Map data Map data | reconstruction of Tulane virus | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Tulane virus Tulane virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.6 Å | |||||||||
 Authors Authors | Yu G / Li K / Jiang W | |||||||||
 Citation Citation | Journal: Structure / Year: 2016 Title: Antibody-Based Affinity Cryoelectron Microscopy at 2.6-Å Resolution. Authors: Guimei Yu / Kunpeng Li / Pengwei Huang / Xi Jiang / Wen Jiang /  Abstract: The affinity cryoelectron microscopy (cryo-EM) approach has been explored in recent years to simplify and/or improve the sample preparation for cryo-EM, which can bring previously challenging ...The affinity cryoelectron microscopy (cryo-EM) approach has been explored in recent years to simplify and/or improve the sample preparation for cryo-EM, which can bring previously challenging specimens such as those of low abundance and/or unpurified ones within reach of the cryo-EM technique. Despite the demonstrated successes for solving structures to low to intermediate resolutions, the lack of near-atomic structures using this approach has led to a common perception of affinity cryo-EM as a niche technique incapable of reaching high resolutions. Here, we report a ∼2.6-Å structure solved using the antibody-based affinity grid approach with low-concentration Tulane virus purified from a low-yield cell-culture system that has been challenging to standard cryo-EM grid preparation. Quantitative analyses of the structure indicate data and reconstruction quality comparable with the conventional grid preparation method using samples at high concentration. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8252.map.gz | 210.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8252-v30.xmlemd-8252.xml | 11.7 KB 11.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8252_fsc.xml | 26.2 KB | Display | FSC data file |
| Images | 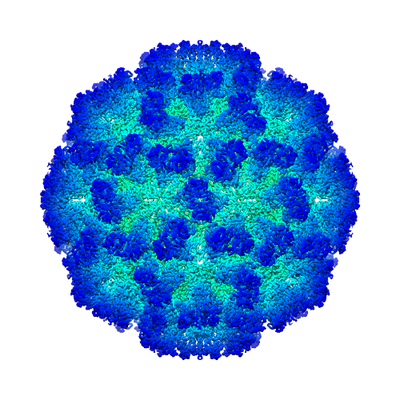 emd_8252.png emd_8252.png | 181.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8252ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8252 http://ftp.pdbj.org/pub/emdb/structures/EMD-8252ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8252 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_8252.map.gz / Format: CCP4 / Size: 1000 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | reconstruction of Tulane virus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.975 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
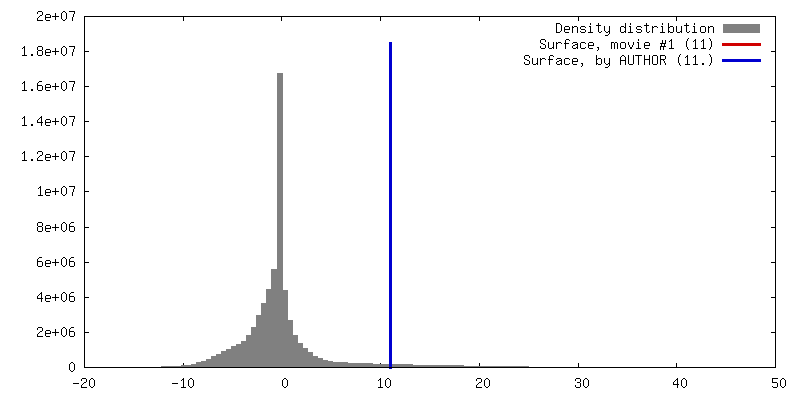
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Tulane virus
| Entire | Name: Tulane virus |
|---|---|
| Components |
|
-Supramolecule #1: Tulane virus
| Supramolecule | Name: Tulane virus / type: virus / ID: 1 / Parent: 0 / Details: Purified from cell cultures / NCBI-ID: 512169 / Sci species name: Tulane virus / Virus type: VIRION / Virus isolate: STRAIN / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Macaca mulatta (Rhesus monkey) Macaca mulatta (Rhesus monkey) |
| Host system | Organism: Platyrrhini (New World monkeys) / Recombinant cell: Monkey kidney cells / Recombinant plasmid: pBR322 |
| Molecular weight | Theoretical: 10 MDa |
| Virus shell | Shell ID: 1 / Diameter: 400.0 Å / T number (triangulation number): 3 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.4 / Details: 1x PBS |
|---|---|
| Grid | Model: Ted Pella ultrathin carbon / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 3.0 nm / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 60 % / Chamber temperature: 298 K / Instrument: GATAN CRYOPLUNGE 3 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal magnification: 22500 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Frames/image: 2-25 / Number grids imaged: 1 / Number real images: 1000 / Average electron dose: 5.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 20000 |
|---|---|
| CTF correction | Software - Name: fitctf2.py Software - details: http://jiang.bio.purdue.edu/software.php Details: Amplitude correction was performed during 3D reconstruction. |
| Startup model | Type of model: OTHER Details: Random initial models were generated using a small fraction of the boxed particles. |
| Initial angle assignment | Type: PROJECTION MATCHING / Software - Name: jspr Software - details: http://jiang.bio.purdue.edu/software.php |
| Final angle assignment | Type: PROJECTION MATCHING / Software - Name: jspr Software - details: http://jiang.bio.purdue.edu/software.php |
| Final reconstruction | Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 2.6 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: jspr Software - details: http://jiang.bio.purdue.edu/software.php Number images used: 14154 |
| FSC plot (resolution estimation) |  |
