 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-7294: Cryo-EM Structure of Hepatitis B virus T=4 capsid in complex with... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7294 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM Structure of Hepatitis B virus T=4 capsid in complex with the fluorescent allosteric modulator HAP-TAMRA | |||||||||
 Map data Map data | A Hepatitis B virus capsid with T=4 symmetry, bound to HAP-TAMRA. HAP-TAMRA is a fluorescent probe derived from the Heteroaryldihydropyrimidine (HAP) class of capsid-directed antivirals. | |||||||||
 Sample Sample | Hepatitis B virus T=4 capsid != Hepatitis B virus genotype D subtype adw Hepatitis B virus T=4 capsid
| |||||||||
| Function / homology |  Function and homology information Function and homology informationmicrotubule-dependent intracellular transport of viral material towards nucleus / T=4 icosahedral viral capsid / viral penetration into host nucleus / host cell cytoplasm / symbiont entry into host cell / structural molecule activity /  DNA binding / RNA binding / identical protein binding DNA binding / RNA binding / identical protein bindingSimilarity search - Function | |||||||||
| Biological species |  Hepatitis B virus genotype D subtype adw Hepatitis B virus genotype D subtype adw | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.0 Å | |||||||||
 Authors Authors | Schlicksup C / Wang JC / Zlotnick A | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2018 Title: Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids. Authors: Christopher John Schlicksup / Joseph Che-Yen Wang / Samson Francis / Balasubramanian Venkatakrishnan / William W Turner / Michael VanNieuwenhze / Adam Zlotnick / Abstract: Defining mechanisms of direct-acting antivirals facilitates drug development and our understanding of virus function. Heteroaryldihydropyrimidines (HAPs) inappropriately activate assembly of ...Defining mechanisms of direct-acting antivirals facilitates drug development and our understanding of virus function. Heteroaryldihydropyrimidines (HAPs) inappropriately activate assembly of hepatitis B virus (HBV) core protein (Cp), suppressing formation of virions. We examined a fluorophore-labeled HAP, HAP-TAMRA. HAP-TAMRA induced Cp assembly and also bound pre-assembled capsids. Kinetic and spectroscopic studies imply that HAP-binding sites are usually not available but are bound cooperatively. Using cryo-EM, we observed that HAP-TAMRA asymmetrically deformed capsids, creating a heterogeneous array of sharp angles, flat regions, and outright breaks. To achieve high resolution reconstruction (<4 Å), we introduced a disulfide crosslink that rescued particle symmetry. We deduced that HAP-TAMRA caused quasi-sixfold vertices to become flatter and fivefold more angular. This transition led to asymmetric faceting. That a disordered crosslink could rescue symmetry implies that capsids have tensegrity properties. Capsid distortion and disruption is a new mechanism by which molecules like the HAPs can block HBV infection. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7294.map.gz | 195.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7294-v30.xmlemd-7294.xml | 13.2 KB 13.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_7294_fsc.xml | 13.1 KB | Display | FSC data file |
| Images |  emd_7294.png emd_7294.png | 212.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7294ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7294 http://ftp.pdbj.org/pub/emdb/structures/EMD-7294ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7294 | HTTPS FTP |
-Related structure data
| Related structure data |  6bvfMC  7295C  6bvnC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_7294.map.gz / Format: CCP4 / Size: 209.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | A Hepatitis B virus capsid with T=4 symmetry, bound to HAP-TAMRA. HAP-TAMRA is a fluorescent probe derived from the Heteroaryldihydropyrimidine (HAP) class of capsid-directed antivirals. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.285 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Hepatitis B virus T=4 capsid
| Entire | Name: Hepatitis B virus T=4 capsid |
|---|---|
| Components |
|
-Supramolecule #1: Hepatitis B virus genotype D subtype adw
| Supramolecule | Name: Hepatitis B virus genotype D subtype adw / type: virus / ID: 1 / Parent: 0 / Macromolecule list: #1 / NCBI-ID: 10419 / Sci species name: Hepatitis B virus genotype D subtype adw / Sci species strain: isolate United Kingdom/adyw/1979 / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: OTHER / Virus enveloped: No / Virus empty: Yes |
|---|---|
| Host system | Organism:  Escherichia coli (E. coli) Escherichia coli (E. coli) |
-Macromolecule #1: Capsid protein
| Macromolecule | Name: Capsid protein / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Hepatitis B virus genotype D subtype adw |
| Molecular weight | Theoretical: 16.791104 KDa |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: MDIDPYKEFG ATVELLSFLP SDFFPSVRDL LDTAAALYRD ALESPEHASP HHTALRQAIL AWGDLMTLAT WVGTNLEDPA SRDLVVSYV NTNVGLKFRQ LLWFHISALT FGRETVLEYL VSFGVWIRTP PAYRPPNAPI LSTLPETTVV C |
-Macromolecule #2: Heteroaryldihydropyrimidine tetramethylrodamine
| Macromolecule | Name: Heteroaryldihydropyrimidine tetramethylrodamine / type: ligand / ID: 2 / Number of copies: 2 / Formula: E9D |
|---|---|
| Molecular weight | Theoretical: 939.428 Da |
| Chemical component information |  ChemComp-E9D: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 10 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| |||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 300 | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 295.15 K / Instrument: FEI VITROBOT MARK III |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Number grids imaged: 1 / Number real images: 679 / Average electron dose: 33.0 e/Å2 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 24823 |
|---|---|
| CTF correction | Software - Name: CTFFIND (ver. 4.1) |
| Startup model | Type of model: NONE / Details: A spherical volume with a diameter of 35 nm |
| Initial angle assignment | Type: NOT APPLICABLE |
| Final angle assignment | Type: NOT APPLICABLE |
| Final reconstruction | Applied symmetry - Point group: I (icosahedral) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 4.0 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 2.1) / Software - details: Automated B-factor Sharpening / Number images used: 16008 |
| FSC plot (resolution estimation) | 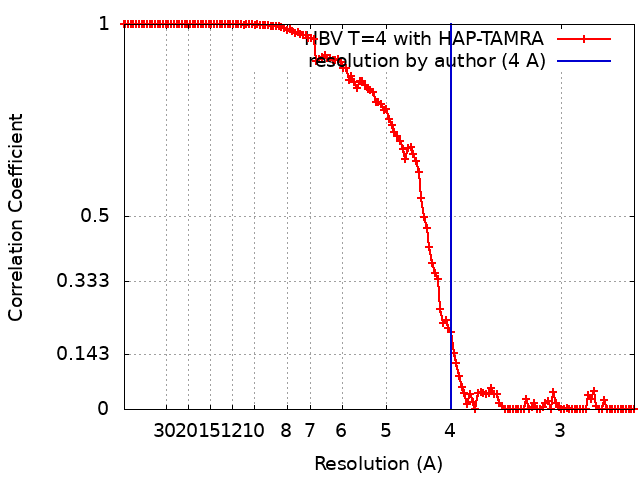 |

