 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7023 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Mitochondrial ATPase Protease YME1 | ||||||||||||||||||||||||
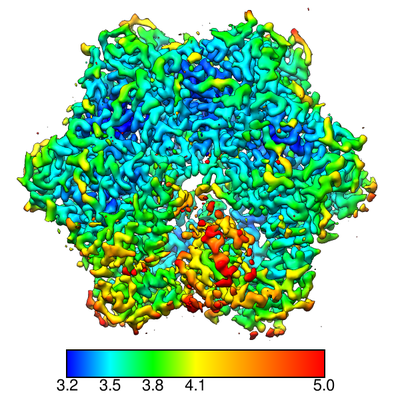
 Map data Map data | sharpened map | ||||||||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||||||||
 Keywords Keywords |  Mitochondrial / ATPase / Protease / HYDROLASE Mitochondrial / ATPase / Protease / HYDROLASE | ||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationATP-dependent peptidase activity / metalloendopeptidase activity / ATP binding / membraneSimilarity search - Function | ||||||||||||||||||||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) / Saccharomyces cerevisiae (strain RM11-1a) (yeast) / Saccharomyces cerevisiae (brewer's yeast) / Saccharomyces cerevisiae (strain RM11-1a) (yeast) /  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.4 Å | ||||||||||||||||||||||||
 Authors Authors | Puchades C / Rampello AJ | ||||||||||||||||||||||||
| Funding support |  United States, 7 items United States, 7 items
| ||||||||||||||||||||||||
 Citation Citation | Journal: Science / Year: 2017 Title: Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Authors: Cristina Puchades / Anthony J Rampello / Mia Shin / Christopher J Giuliano / R Luke Wiseman / Steven E Glynn / Gabriel C Lander / Abstract: We present an atomic model of a substrate-bound inner mitochondrial membrane AAA+ quality control protease in yeast, YME1. Our ~3.4-angstrom cryo-electron microscopy structure reveals how the ...We present an atomic model of a substrate-bound inner mitochondrial membrane AAA+ quality control protease in yeast, YME1. Our ~3.4-angstrom cryo-electron microscopy structure reveals how the adenosine triphosphatases (ATPases) form a closed spiral staircase encircling an unfolded substrate, directing it toward the flat, symmetric protease ring. Three coexisting nucleotide states allosterically induce distinct positioning of tyrosines in the central channel, resulting in substrate engagement and translocation to the negatively charged proteolytic chamber. This tight coordination by a network of conserved residues defines a sequential, around-the-ring adenosine triphosphate hydrolysis cycle that results in stepwise substrate translocation. A hingelike linker accommodates the large-scale nucleotide-driven motions of the ATPase spiral relative to the planar proteolytic base. The translocation mechanism is likely conserved for other AAA+ ATPases. | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7023.map.gz | 11.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7023-v30.xmlemd-7023.xml | 33.6 KB 33.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_7023_fsc.xml | 9 KB | Display | FSC data file |
| Images |  emd_7023.png emd_7023.png | 199.9 KB | ||
| Filedesc metadata | emd-7023.cif.gz | 7.1 KB | ||
| Others | emd_7023_additional_1.map.gzemd_7023_additional_2.map.gzemd_7023_additional_3.map.gzemd_7023_half_map_1.map.gzemd_7023_half_map_2.map.gzemd_7023_half_map_3.map.gzemd_7023_half_map_4.map.gz | 1.7 MB 1.6 MB 4.4 MB 1.6 MB 1.6 MB 4.4 MB 4.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7023ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7023 http://ftp.pdbj.org/pub/emdb/structures/EMD-7023ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7023 | HTTPS FTP |
-Related structure data
| Related structure data |  6az0MC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_7023.map.gz / Format: CCP4 / Size: 12.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | sharpened map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.03 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
-Additional map: focused refinement of step subunit, unsharpened
| File | emd_7023_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | focused refinement of step subunit, unsharpened | ||||||||||||
| Projections & Slices |
| ||||||||||||
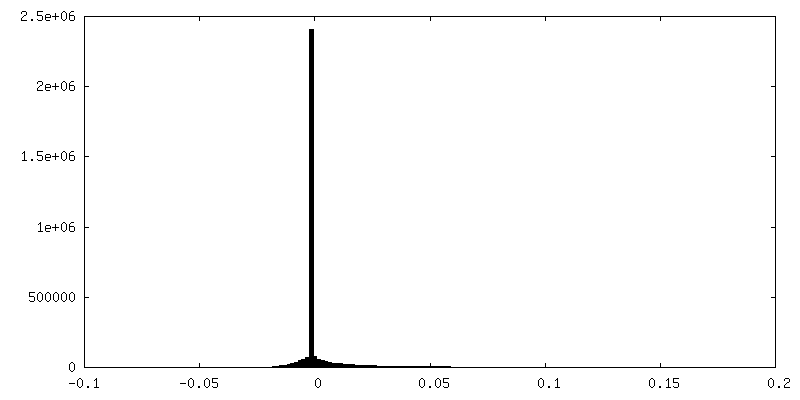
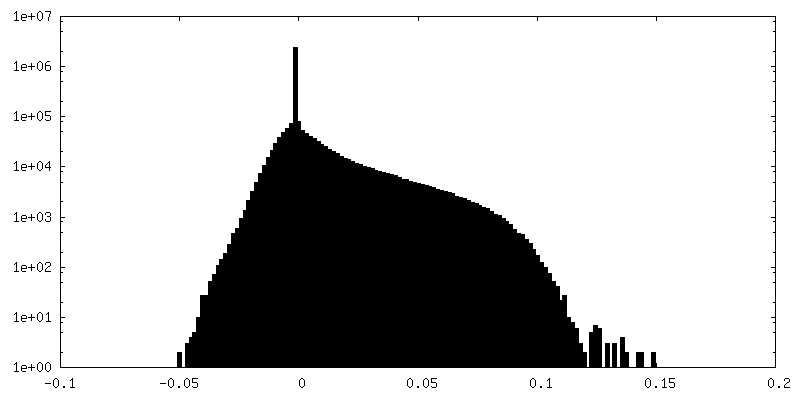
| Density Histograms |
 Z
Z Y
Y X
X





-Additional map: focused refinement of step subunit, sharpened
| File | emd_7023_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | focused refinement of step subunit, sharpened | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Additional map: unsharpened map
| File | emd_7023_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | unsharpened map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


-Half map: focused refinement of step subunit, half map 2
| File | emd_7023_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | focused refinement of step subunit, half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




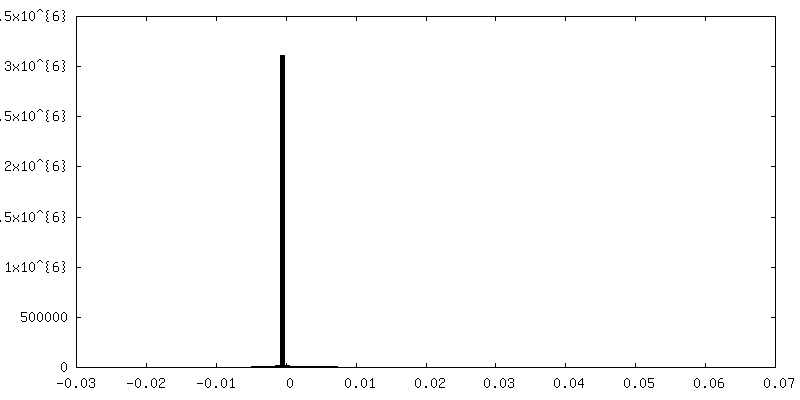

-Half map: focused refinement of step subunit, half map 1
| File | emd_7023_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | focused refinement of step subunit, half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




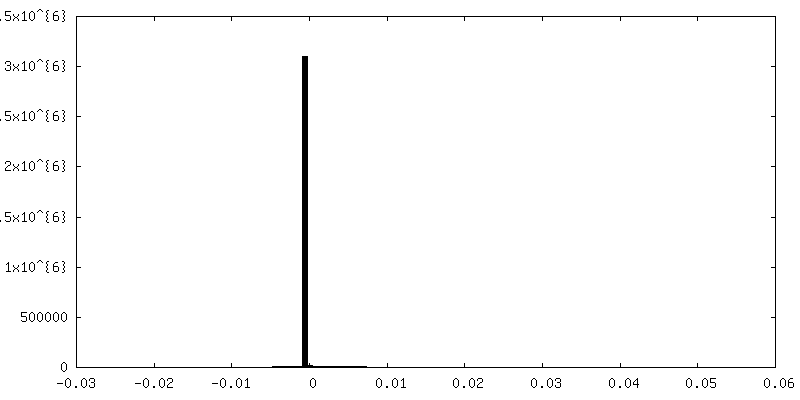
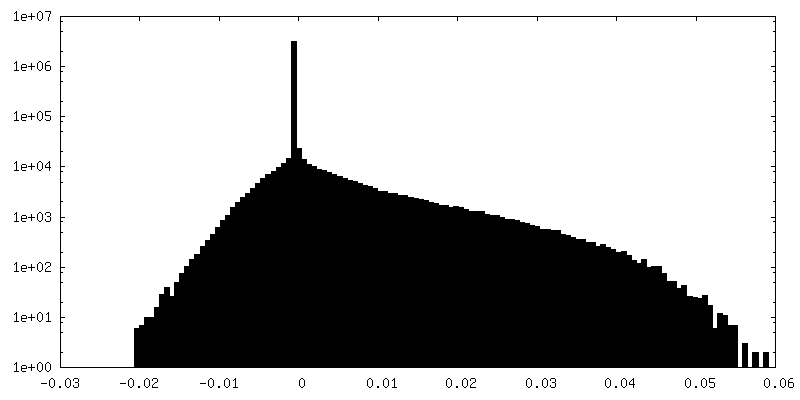
-Half map: half map 1
| File | emd_7023_half_map_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







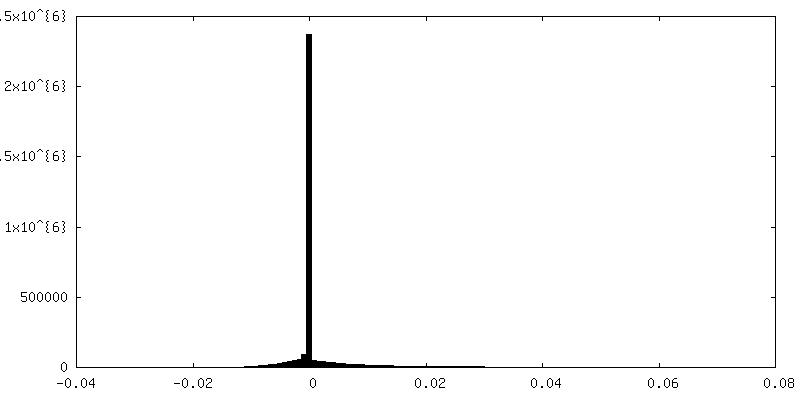
-Half map: half map 2
| File | emd_7023_half_map_4.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : YME1 ATPase Protease Walker B mutant bound to substrate
| Entire | Name: YME1 ATPase Protease Walker B mutant bound to substrate |
|---|---|
| Components |
|
-Supramolecule #1: YME1 ATPase Protease Walker B mutant bound to substrate
| Supramolecule | Name: YME1 ATPase Protease Walker B mutant bound to substrate type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 Details: YME1 Walker B mutant was recombinantly expressed in E. coli, solved in presence of ATP with unknown bound substrate. |
|---|---|
| Source (natural) | Organism: Saccharomyces cerevisiae (brewer's yeast) |
| Molecular weight | Theoretical: 295 KDa |
-Macromolecule #1: Mitochondrial inner membrane i-AAA protease supercomplex subunit YME1
| Macromolecule | Name: Mitochondrial inner membrane i-AAA protease supercomplex subunit YME1 type: protein_or_peptide / ID: 1 / Details: Walker B mutant / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Saccharomyces cerevisiae (strain RM11-1a) (yeast) / Strain: RM11-1a |
| Molecular weight | Theoretical: 48.025758 KDa |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: KFDDVCGCDE ARAELEEIVD FLKDPTKYES LGGKLPKGVL LTGPPGTGKT LLARATAGEA GVDFFFMSGS EFDEVYVGVG AKRIRDLFA QARSRAPAII FIDQLDAIGG KRNPKDQAYA KQTLNQLLVE LDGFSQTSGI IIIGATNFPE ALDKALTRPG R FDKVVNVD ...String: KFDDVCGCDE ARAELEEIVD FLKDPTKYES LGGKLPKGVL LTGPPGTGKT LLARATAGEA GVDFFFMSGS EFDEVYVGVG AKRIRDLFA QARSRAPAII FIDQLDAIGG KRNPKDQAYA KQTLNQLLVE LDGFSQTSGI IIIGATNFPE ALDKALTRPG R FDKVVNVD LPDVRGRADI LKHHMKKITL ADNVDPTIIA RGTPGLSGAE LANLVNQAAV YACQKNAVSV DMSHFEWAKD KI LMGAERK TMVLTDAARK ATAFHEAGHA IMAKYTNGAT PLYKATILPR GRALGITFQL PEMDKVDITK RECQARLDVC MGG KIAEEL IYGKDNTTSG CGSDLQSATG TARAMVTQYG MSDDVGPVNL SEEWESWSNK IRDIADNEVI ELLKDSEERA RRLL TKKNV ELHRLAQGLI EYETLDAHEI EQVCKGEKLA KLKT UniProtKB: AAA domain-containing protein |
-Macromolecule #2: poly(UNK)
| Macromolecule | Name: poly(UNK) / type: protein_or_peptide / ID: 2 / Details: unknown polypeptide / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Escherichia coli (E. coli) |
| Molecular weight | Theoretical: 869.063 Da |
| Sequence | String: (UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK) |
-Macromolecule #3: ADENOSINE-5'-TRIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-TRIPHOSPHATE / type: ligand / ID: 3 / Number of copies: 4 / Formula: ATP |
|---|---|
| Molecular weight | Theoretical: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-Macromolecule #4: ZINC ION
| Macromolecule | Name: ZINC ION / type: ligand / ID: 4 / Number of copies: 6 / Formula: ZN |
|---|---|
| Molecular weight | Theoretical: 65.409 Da |
-Macromolecule #5: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 5 / Number of copies: 4 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Macromolecule #6: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 6 / Number of copies: 1 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 8 Component:
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grid | Model: Quantifoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Time: 6 sec. / Pretreatment - Atmosphere: OTHER / Details: Gatan Solarus Plasma Cleaner | |||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated defocus max: 2.5 µm / Calibrated defocus min: 1.2 µm / Calibrated magnification: 51500 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 29000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Temperature | Min: 70.0 K / Max: 70.0 K |
| Details | Data collected using Leginon |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Frames/image: 1-35 / Number grids imaged: 3 / Number real images: 6098 / Average exposure time: 7.0 sec. / Average electron dose: 60.0 e/Å2 Details: Images collected in counting mode at 5 frames per second. |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 2285499 Details: Particles picked automatically with FindEM template picking |
|---|---|
| Startup model | Type of model: INSILICO MODEL In silico model: common lines model generated from negative stain data |
| Initial angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) / Details: RELION 1.4 |
| Final angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) / Details: RELION 1.4 |
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Algorithm: BACK PROJECTION / Resolution.type: BY AUTHOR / Resolution: 3.4 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 1.4) / Number images used: 62917 |
| FSC plot (resolution estimation) |  |
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: AB INITIO MODEL / Overall B value: 100 |
|---|---|
| Output model | PDB-6az0: |
