 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
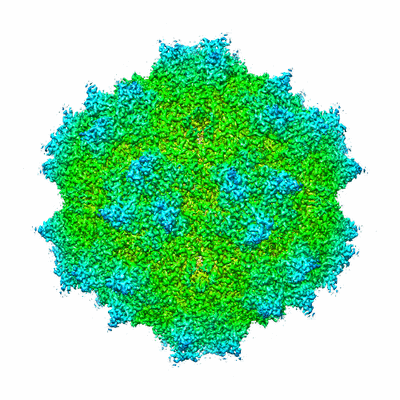
Yorodumi- EMDB-6470: Single particle reconstruction of AAV-DJ in complex with ARIXTRA -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-6470 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Single particle reconstruction of AAV-DJ in complex with ARIXTRA | |||||||||
 Map data Map data | Reconstruction of adeno-associated virus variant DJ in complex with Arixtra | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords |  gene therapy gene therapy | |||||||||
| Biological species | unidentified (others) /   Adeno-associated virus Adeno-associated virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.8 Å | |||||||||
 Authors Authors | Spear JM / Noble AJ / Xie Q / Sousa DR / Chapman MS / Stagg SM | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2015 Title: The influence of frame alignment with dose compensation on the quality of single particle reconstructions. Authors: John M Spear / Alex J Noble / Qing Xie / Duncan R Sousa / Michael S Chapman / Scott M Stagg /  Abstract: As direct electron detection devices in cryo-electron microscopy become ubiquitous, the field is now ripe for new developments in image analysis techniques that take advantage of their increased SNR ...As direct electron detection devices in cryo-electron microscopy become ubiquitous, the field is now ripe for new developments in image analysis techniques that take advantage of their increased SNR coupled with their high-throughput frame collection abilities. In approaching atomic resolution of native-like biomolecules, the accurate extraction of structural locations and orientations of side-chains from frames depends not only on the electron dose that a sample receives but also on the ability to accurately estimate the CTF. Here we use a new 2.8Å resolution structure of a recombinant gene therapy virus, AAV-DJ with Arixtra, imaged on an FEI Titan Krios with a DE-20 direct electron detector to probe new metrics including relative side-chain density and ResLog analysis for optimizing the compensation of electron beam damage and to characterize the factors that are limiting the resolution of the reconstruction. The influence of dose compensation on the accuracy of CTF estimation and particle classifiability are also presented. We show that rigorous dose compensation allows for better particle classifiability and greater recovery of structural information from negatively charged, electron-sensitive side-chains, resulting in a more accurate macromolecular model. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_6470.map.gz | 258.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-6470-v30.xmlemd-6470.xml | 11.2 KB 11.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_6470_fsc.xml | 17.7 KB | Display | FSC data file |
| Images |  400_6470.gif 400_6470.gif 80_6470.gif 80_6470.gif | 90.7 KB 4.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-6470ftp://ftp.pdbj.org/pub/emdb/structures/EMD-6470 http://ftp.pdbj.org/pub/emdb/structures/EMD-6470ftp://ftp.pdbj.org/pub/emdb/structures/EMD-6470 | HTTPS FTP |
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_6470.map.gz / Format: CCP4 / Size: 300.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of adeno-associated virus variant DJ in complex with Arixtra | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.215 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Adeno-associated virus
| Entire | Name: Adeno-associated virus |
|---|---|
| Components |
|
-Supramolecule #1000: Adeno-associated virus
| Supramolecule | Name: Adeno-associated virus / type: sample / ID: 1000 Oligomeric state: 60 viral subunits form the icosahedral capsid Number unique components: 2 |
|---|---|
| Molecular weight | Theoretical: 3.75 MDa |
-Supramolecule #1: Adeno-associated virus
| Supramolecule | Name: Adeno-associated virus / type: virus / ID: 1 / NCBI-ID: 272636 / Sci species name: Adeno-associated virus / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: SEROTYPE / Virus enveloped: No / Virus empty: Yes / Sci species serotype: DJ |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) / synonym: VERTEBRATES Homo sapiens (human) / synonym: VERTEBRATES |
| Host system | Organism:   Spodoptera frugiperda (fall armyworm) / Recombinant cell: SF9 / Recombinant plasmid: bacmid Spodoptera frugiperda (fall armyworm) / Recombinant cell: SF9 / Recombinant plasmid: bacmid |
| Molecular weight | Experimental: 3.75 MDa |
| Virus shell | Shell ID: 1 / Name: Shell 1 / Diameter: 250 Å / T number (triangulation number): 1 |
-Macromolecule #1: Fondaparinux
| Macromolecule | Name: Fondaparinux / type: ligand / ID: 1 / Name.synonym: Arixtra Details: Arixtra was added in a 10X molar excess to AAV-DJ monomers Recombinant expression: No / Database: NCBI |
|---|---|
| Source (natural) | Organism: unidentified (others) |
| Molecular weight | Experimental: 1.727 KDa |
| Chemical component information | 
ChemComp-PRD_900028: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.6 mg/mL |
|---|---|
| Buffer | pH: 7.4 / Details: 50mM HEPES, 25mM MgCl2, 25mM NaCl |
| Grid | Details: R2/2 200 mesh copper grids that were rendered hydrophilic in 75/25 % Ar/O. |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 120 K / Instrument: FEI VITROBOT MARK IV Details: 3ul of 0.6 mg/ml AAV-DJ was applied to grid and then hand blotted. After initial hand blotting, 3ul of 5.7mM Arixtra was added, the grid was mechanically blotted, and then vitrified. Method: blot force = 1, blot time = 3 seconds, total blots = 1. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 49383 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 0.75 µm / Nominal magnification: 29000 |
| Sample stage | Specimen holder: liquid nitrogen cooled / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Temperature | Average: 93 K |
| Alignment procedure | Legacy - Astigmatism: Astigmatism was corrected at 155000 times magnification |
| Date | Jan 6, 2015 |
| Image recording | Category: CCD / Film or detector model: DIRECT ELECTRON DE-20 (5k x 3k) / Number real images: 1051 / Average electron dose: 66 e/Å2 / Details: every image is the sum of 45 frames / Bits/pixel: 16 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| CTF correction | Details: by image |
|---|---|
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 2.8 Å / Resolution method: OTHER / Software - Name: EMAN, Frealign / Number images used: 120166 |
| Details | Particles were picked using an automatic selection program. Particles over carbon were manually deselected. |
| FSC plot (resolution estimation) | 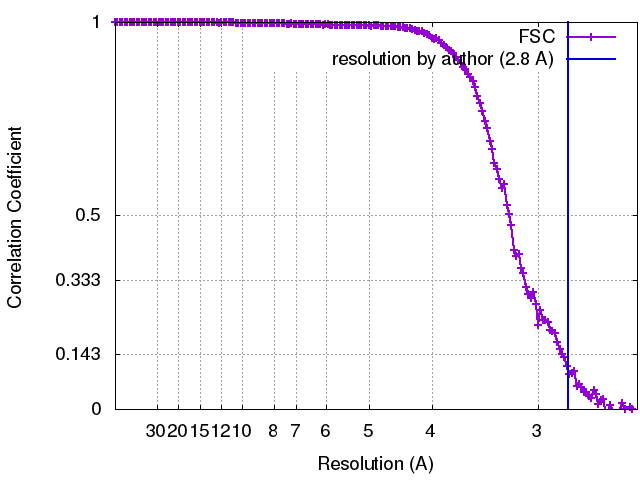 |
