 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-5952 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
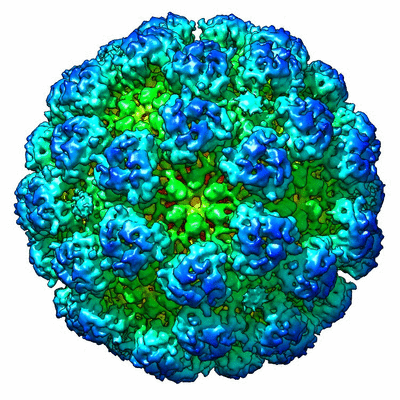
| Title | CryoEM reconstruction model of Orsay virus-like particle | |||||||||
 Map data Map data | Reconstruction of recombinant Orsay virus-like particle | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords |  virus-like particle virus-like particle | |||||||||
| Function / homology | Nodavirus capsid / nodavirus capsid protein / Viral coat protein subunit / metal ion binding / Capsid protein alpha Function and homology information Function and homology information | |||||||||
| Biological species |  Orsay virus Orsay virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.9 Å | |||||||||
 Authors Authors | Guo YR / Hryc C / Jakana J / Jiang H / Wang D / Chiu W / Zhong W / Tao YJ | |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2014 Title: Crystal structure of a nematode-infecting virus. Authors: Yusong R Guo / Corey F Hryc / Joanita Jakana / Hongbing Jiang / David Wang / Wah Chiu / Weiwei Zhong / Yizhi J Tao /  Abstract: Orsay, the first virus discovered to naturally infect Caenorhabditis elegans or any nematode, has a bipartite, positive-sense RNA genome. Sequence analyses show that Orsay is related to nodaviruses, ...Orsay, the first virus discovered to naturally infect Caenorhabditis elegans or any nematode, has a bipartite, positive-sense RNA genome. Sequence analyses show that Orsay is related to nodaviruses, but molecular characterizations of Orsay reveal several unique features, such as the expression of a capsid-δ fusion protein and the use of an ATG-independent mechanism for translation initiation. Here we report the crystal structure of an Orsay virus-like particle assembled from recombinant capsid protein (CP). Orsay capsid has a T = 3 icosahedral symmetry with 60 trimeric surface spikes. Each CP can be divided into three regions: an N-terminal arm that forms an extended protein interaction network at the capsid interior, an S domain with a jelly-roll, β-barrel fold forming the continuous capsid, and a P domain that forms surface spike projections. The structure of the Orsay S domain is best aligned to T = 3 plant RNA viruses but exhibits substantial differences compared with the insect-infecting alphanodaviruses, which also lack the P domain in their CPs. The Orsay P domain is remotely related to the P1 domain in calicivirus and hepatitis E virus, suggesting a possible evolutionary relationship. Removing the N-terminal arm produced a slightly expanded capsid with fewer nucleic acids packaged, suggesting that the arm is important for capsid stability and genome packaging. Because C. elegans-Orsay serves as a highly tractable model for studying viral pathogenesis, our results should provide a valuable structural framework for further studies of Orsay replication and infection. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_5952.map.gz | 12.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-5952-v30.xmlemd-5952.xml | 12.9 KB 12.9 KB | Display Display | EMDB header |
| Images |  400_5952.gif 400_5952.gif 80_5952.gif 80_5952.gif | 109.4 KB 6.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-5952ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5952 http://ftp.pdbj.org/pub/emdb/structures/EMD-5952ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5952 | HTTPS FTP |
-Related structure data










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_5952.map.gz / Format: CCP4 / Size: 29.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of recombinant Orsay virus-like particle | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.15 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : recombinant virus-like particle of Orsay virus
| Entire | Name: recombinant virus-like particle of Orsay virus |
|---|---|
| Components |
|
-Supramolecule #1000: recombinant virus-like particle of Orsay virus
| Supramolecule | Name: recombinant virus-like particle of Orsay virus / type: sample / ID: 1000 / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 7.7 MDa |
-Supramolecule #1: Orsay virus
| Supramolecule | Name: Orsay virus / type: virus / ID: 1 / Name.synonym: Orsay virus-like particle / NCBI-ID: 977912 / Sci species name: Orsay virus / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: SPECIES / Virus enveloped: No / Virus empty: No / Syn species name: Orsay virus-like particle |
|---|---|
| Host (natural) | Organism:  Caenorhabditis elegans (invertebrata) / synonym: INVERTEBRATES Caenorhabditis elegans (invertebrata) / synonym: INVERTEBRATES |
| Host system | Organism:  Escherichia coli (E. coli) / Recombinant strain: Rosetta / Recombinant plasmid: pET28a Escherichia coli (E. coli) / Recombinant strain: Rosetta / Recombinant plasmid: pET28a |
| Molecular weight | Theoretical: 7.2 MDa |
| Virus shell | Shell ID: 1 / Name: Orsay VLP Capsid Protein / Diameter: 360 Å / T number (triangulation number): 3 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 7.5 Details: 50 mM Tris, 500 mM NaCl, 2 mM EDTA, 5 mM 2-mercaptoethanol |
| Grid | Details: 400 mesh Quantifoil R 1.2/1.3 copper grids with thin carbon support |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 80 K / Instrument: FEI VITROBOT MARK III / Method: Blot for 2 seconds before plunging |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 3200FSC |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Nominal defocus max: 4.0 µm / Nominal defocus min: 1.3 µm / Nominal magnification: 25000 |
| Specialist optics | Energy filter - Name: in-column filter / Energy filter - Lower energy threshold: 0.0 eV / Energy filter - Upper energy threshold: 25.0 eV |
| Sample stage | Specimen holder model: JEOL 3200FSC CRYOHOLDER |
| Temperature | Average: 81 K |
| Date | Oct 2, 2012 |
| Image recording | Category: CCD / Film or detector model: DIRECT ELECTRON DE-12 (4k x 3k) / Digitization - Sampling interval: 6 µm / Number real images: 112 / Average electron dose: 25 e/Å2 Details: Each image is the average of subframes 2-36 after drift and damage correction. Bits/pixel: 10 |
-Image processing
| CTF correction | Details: Particles per frame |
|---|---|
| Final two d classification | Number classes: 25 |
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 6.9 Å / Resolution method: OTHER / Software - Name: EMAN2 Details: The final map was generated from two independent data sets. Small subsets (~200 particles) of each data set were extracted from each half of the data and binned by 4X. Two independent ...Details: The final map was generated from two independent data sets. Small subsets (~200 particles) of each data set were extracted from each half of the data and binned by 4X. Two independent initial models were generated using EMAN2. The remaining particles were added to the two data sets and then refined independently (not binned) with icosahedral symmetry enforced. Angular step size was reduced after 15 iterations; after 25 iterations of refinement the data sets converged to a final density map. A low-pass filter was applied to the capsid protein portion of the density map and a soft-edge mask was used to remove the non-icosahedral portion of the density map (RNA). Gold Standard resolution assessment resulted in a final Fourier shell correlation (FSC) of 6.9A resolution between the two independent density maps, using the 0.143 criterion. The final density map was generated by combining the two data sets and performing one last round of refinement. Number images used: 4332 |
| Details | 112 images were imported into EMAN2, evaluated, and frames with a high amount of contamination and/or ice were removed. A total of 5,824 particles were boxed out (300 x 300 pixels) from the raw images using a semi-automated routine. Damage and drift correction done on the individual particles showed that minimal drift (<2A) had occurred for the complete data set. The corrected particles were imported back into EMAN2 and the contrast transfer function (CTF) was determined automatically followed by manual correction. Defocus was estimated to be between 1.3 and 4 um. Various subsets of particles were removed due on poor CTF parameters, resulting in the 4,332 particles that were used in the reconstruction. |
-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Software | Name: Chimera |
| Details | The molecular model was determined by X-ray crystallography using this cryo-EM density map to phase the crystal. The model was then fit back into the density map with Chimera. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
