 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3573 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
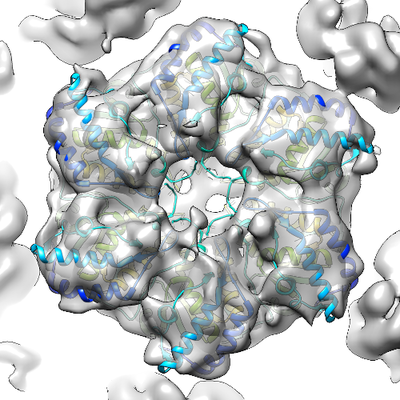
| Title | Localized reconstruction of bacteriophage phi6 vertex | |||||||||
 Map data Map data | Localized reconstruction of bacteriophage phi6 vertex | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationviral procapsid / viral genome packaging / ribonucleoside triphosphate phosphatase activity /  viral capsid / nucleoside-triphosphate phosphatase / ATP binding viral capsid / nucleoside-triphosphate phosphatase / ATP bindingSimilarity search - Function | |||||||||
| Biological species |  Pseudomonas phage phi6 (bacteriophage) Pseudomonas phage phi6 (bacteriophage) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 9.1 Å | |||||||||
 Authors Authors | Sun Z / El Omari K / Sun X / Ilca S / Kotecha A / Stuart DI / Poranen MM / Huiskonen JT | |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2013 Title: Tracking in atomic detail the functional specializations in viral RecA helicases that occur during evolution. Authors: Kamel El Omari / Christoph Meier / Denis Kainov / Geoff Sutton / Jonathan M Grimes / Minna M Poranen / Dennis H Bamford / Roman Tuma / David I Stuart / Erika J Mancini /  Abstract: Many complex viruses package their genomes into empty protein shells and bacteriophages of the Cystoviridae family provide some of the simplest models for this. The cystoviral hexameric NTPase, P4, ...Many complex viruses package their genomes into empty protein shells and bacteriophages of the Cystoviridae family provide some of the simplest models for this. The cystoviral hexameric NTPase, P4, uses chemical energy to translocate single-stranded RNA genomic precursors into the procapsid. We previously dissected the mechanism of RNA translocation for one such phage, 12, and have now investigated three further highly divergent, cystoviral P4 NTPases (from 6, 8 and 13). High-resolution crystal structures of the set of P4s allow a structure-based phylogenetic analysis, which reveals that these proteins form a distinct subfamily of the RecA-type ATPases. Although the proteins share a common catalytic core, they have different specificities and control mechanisms, which we map onto divergent N- and C-terminal domains. Thus, the RNA loading and tight coupling of NTPase activity with RNA translocation in 8 P4 is due to a remarkable C-terminal structure, which wraps right around the outside of the molecule to insert into the central hole where RNA binds to coupled L1 and L2 loops, whereas in 12 P4, a C-terminal residue, serine 282, forms a specific hydrogen bond to the N7 of purines ring to confer purine specificity for the 12 enzyme. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3573.map.gz | 7.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3573-v30.xmlemd-3573.xml | 15.9 KB 15.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3573_fsc.xml | 4.5 KB | Display | FSC data file |
| Images |  emd_3573.png emd_3573.png | 214.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3573ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3573 http://ftp.pdbj.org/pub/emdb/structures/EMD-3573ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3573 | HTTPS FTP |
-Related structure data
| Related structure data |  5muwMC  3571C  3572C  5muuC  5muvC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3573.map.gz / Format: CCP4 / Size: 8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Localized reconstruction of bacteriophage phi6 vertex | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Pseudomonas phage phi6
| Entire | Name: Pseudomonas phage phi6 (bacteriophage) |
|---|---|
| Components |
|
-Supramolecule #1: Pseudomonas phage phi6
| Supramolecule | Name: Pseudomonas phage phi6 / type: virus / ID: 1 / Parent: 0 / Macromolecule list: #1 / NCBI-ID: 10879 / Sci species name: Pseudomonas phage phi6 / Virus type: VIRION / Virus isolate: SPECIES / Virus enveloped: Yes / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Pseudomonas syringae (bacteria) Pseudomonas syringae (bacteria) |
-Macromolecule #1: Packaging enzyme P4
| Macromolecule | Name: Packaging enzyme P4 / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO / EC number: nucleoside-triphosphate phosphatase |
|---|---|
| Source (natural) | Organism: Pseudomonas phage phi6 (bacteriophage) |
| Molecular weight | Theoretical: 32.67874 KDa |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: MPIVVTQAHI DRVGIAADLL DASPVSLQVL GRPTAINTVV IKTYIAAVME LASKQGGSLA GVDIRPSVLL KDTAIFTKPK AKSADVESD VDVLDTGIYS VPGLARKPVT HRWPSEGIYS GVTALMGATG SGKSITLNEK LRPDVLIRWG EVAEAYDELD T AVHISTLD ...String: MPIVVTQAHI DRVGIAADLL DASPVSLQVL GRPTAINTVV IKTYIAAVME LASKQGGSLA GVDIRPSVLL KDTAIFTKPK AKSADVESD VDVLDTGIYS VPGLARKPVT HRWPSEGIYS GVTALMGATG SGKSITLNEK LRPDVLIRWG EVAEAYDELD T AVHISTLD EMLIVCIGLG ALGFNVAVDS VRPLLFRLKG AASAGGIVAV FYSLLTDISN LFTQYDCSVV MVVNPMVDAE KI EYVFGQV MASTVGAILC ADGNVSRTMF RTNKGRIFNG AAPLAADTHM PSMDRPTSMK ALDHTSIASV AP |
-Macromolecule #2: CALCIUM ION
| Macromolecule | Name: CALCIUM ION / type: ligand / ID: 2 / Number of copies: 6 / Formula: CA |
|---|---|
| Molecular weight | Theoretical: 40.078 Da |
-Macromolecule #3: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 3 / Number of copies: 6 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3 mg/mL |
|---|---|
| Buffer | pH: 7.2 |
| Grid | Model: C-flat / Material: COPPER / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated defocus max: 3.0 µm / Calibrated defocus min: 0.3 µm / Calibrated magnification: 37037 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.0 mm |
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Lower energy threshold: 0 eV / Energy filter - Upper energy threshold: 20 eV |
| Sample stage | Specimen holder model: OTHER / Cooling holder cryogen: NITROGEN |
| Temperature | Min: 80.0 K / Max: 120.0 K |
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-22 / Number real images: 900 / Average exposure time: 0.2 sec. / Average electron dose: 0.7 e/Å2 |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
-Image processing
| Particle selection | Number selected: 159492 |
|---|---|
| CTF correction | Software: (Name: CTFFIND (ver. 3), RELION (ver. 1.4)) |
| Initial angle assignment | Type: OTHER / Software - Version: 1.1.0 / Software - details: LocalizedReconstruction Details: Angles for P4 hexameter sub-particles were calculated from particle orientations |
| Final 3D classification | Number classes: 6 / Software - Name: RELION (ver. 1.4) |
| Final angle assignment | Type: OTHER |
| Final reconstruction | Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 9.1 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 1.4) / Number images used: 56448 |
| FSC plot (resolution estimation) | 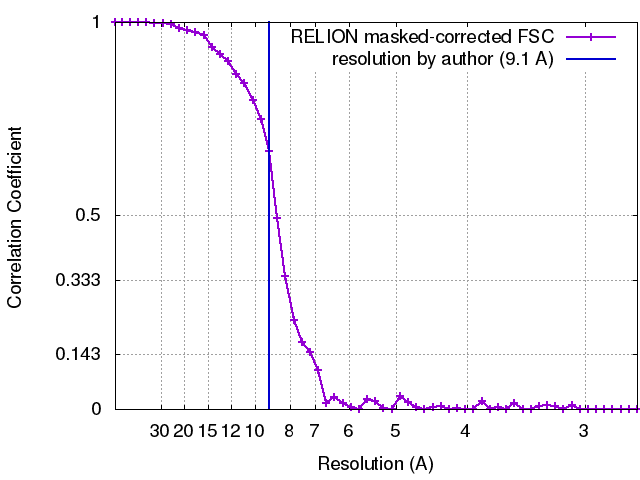 |
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Cross-correlation coefficient |
|---|---|
| Output model | PDB-5muw: |
