 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3450: Structure of the K+ transporter KtrAB from Vibrio alginolyticus i... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3450 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the K+ transporter KtrAB from Vibrio alginolyticus in the ADP-bound state | |||||||||
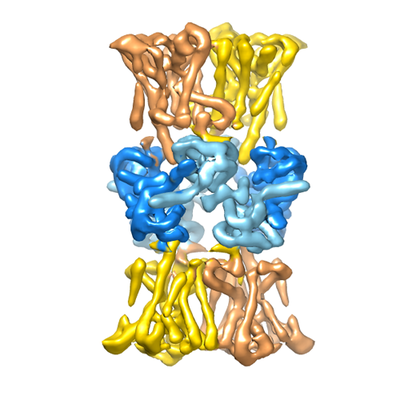
 Map data Map data | KtrB2-KtrA8-KtrB2 complex | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Vibrio alginolyticus (bacteria) Vibrio alginolyticus (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.6 Å | |||||||||
 Authors Authors | Diskowski M / Mills DJ / Baerland N / Haenelt I / Vonck J | |||||||||
 Citation Citation | Journal: Elife / Year: 2017 Title: Helical jackknives control the gates of the double-pore K uptake system KtrAB. Authors: Marina Diskowski / Ahmad Reza Mehdipour / Dorith Wunnicke / Deryck J Mills / Vedrana Mikusevic / Natalie Bärland / Jan Hoffmann / Nina Morgner / Heinz-Jürgen Steinhoff / Gerhard Hummer / ...Authors: Marina Diskowski / Ahmad Reza Mehdipour / Dorith Wunnicke / Deryck J Mills / Vedrana Mikusevic / Natalie Bärland / Jan Hoffmann / Nina Morgner / Heinz-Jürgen Steinhoff / Gerhard Hummer / Janet Vonck / Inga Hänelt /  Abstract: Ion channel gating is essential for cellular homeostasis and is tightly controlled. In some eukaryotic and most bacterial ligand-gated K channels, RCK domains regulate ion fluxes. Until now, a single ...Ion channel gating is essential for cellular homeostasis and is tightly controlled. In some eukaryotic and most bacterial ligand-gated K channels, RCK domains regulate ion fluxes. Until now, a single regulatory mechanism has been proposed for all RCK-regulated channels, involving signal transduction from the RCK domain to the gating area. Here, we present an inactive ADP-bound structure of KtrAB from , determined by cryo-electron microscopy, which, combined with EPR spectroscopy and molecular dynamics simulations, uncovers a novel regulatory mechanism for ligand-induced action at a distance. Exchange of activating ATP to inactivating ADP triggers short helical segments in the K-translocating KtrB dimer to organize into two long helices that penetrate deeply into the regulatory RCK domains, thus connecting nucleotide-binding sites and ion gates. As KtrAB and its homolog TrkAH have been implicated as bacterial pathogenicity factors, the discovery of this functionally relevant inactive conformation may advance structure-guided drug development. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3450.map.gz | 25.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3450-v30.xmlemd-3450.xml | 15.6 KB 15.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3450_fsc.xml | 6.8 KB | Display | FSC data file |
| Images |  emd_3450.png emd_3450.png | 131.9 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3450ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3450 http://ftp.pdbj.org/pub/emdb/structures/EMD-3450ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3450 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3450.map.gz / Format: CCP4 / Size: 27 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | KtrB2-KtrA8-KtrB2 complex | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.67 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : KtrAB
| Entire | Name: KtrAB |
|---|---|
| Components |
|
-Supramolecule #1: KtrAB
| Supramolecule | Name: KtrAB / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: complex of KtrB2A8 with another KtrB transporter dimer attached to the symmetric A8 regulatory domain |
|---|---|
| Source (natural) | Organism: Vibrio alginolyticus (bacteria) |
| Recombinant expression | Organism: Escherichia coli (E. coli) / Recombinant plasmid: pBAD18 |
| Molecular weight | Theoretical: 340 KDa |
-Macromolecule #1: KtrB
| Macromolecule | Name: KtrB / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Vibrio alginolyticus (bacteria) |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: MTQFHQRGVF YVPDGKRDKA KGGEPRIILL SFLGVLLPSA VLLTLPVFSV SGLSITDALF TATSAISVT GLGVVDTGQH FTLAGKILLM CLMQIGGLGQ MTLSAVLLYM FGVRLSLRQQ A LAKEALGQ ERQVNLRRLV KKIVTFALVA EAIGFVFLSY RWVPEMGWQT ...String: MTQFHQRGVF YVPDGKRDKA KGGEPRIILL SFLGVLLPSA VLLTLPVFSV SGLSITDALF TATSAISVT GLGVVDTGQH FTLAGKILLM CLMQIGGLGQ MTLSAVLLYM FGVRLSLRQQ A LAKEALGQ ERQVNLRRLV KKIVTFALVA EAIGFVFLSY RWVPEMGWQT GMFYALFHSI SA FNNAGFA LFSDSMMSFV NDPLVSFTLA GLFIFGGLGF TVIGDVWRHW RKGFHFLHIH TKI MLIATP LLLLVGTVLF WLLERHNPNT MGSLTTGGQW LAAFFQSASA RTAGFNSVDL TQFT QPALL IMIVLMLIGA GSTSTGGGIK VSTFAVAFMA TWTFLRQKKH VVMFKRTVNW PTVTK SLAI IVVSGAILTT AMFLLMLTEK ASFDKVMFET ISAFATVGLT AGLTAELSEP GKYIMI VVM IIGRIGPLTL AYMLARPEPT LIKYPEDTVL TG |
-Macromolecule #2: KtrA
| Macromolecule | Name: KtrA / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Vibrio alginolyticus (bacteria) |
| Recombinant expression | Organism: Escherichia coli (E. coli) |
| Sequence | String: MKTGDKQFAV IGLGRFGLAV CKELQDSGSQ VLAVDINEDR VKEAAGFVSQ AIVANCTHEE TVAELKLDD YDMVMIAIGA DVNASILATL IAKEAGVKSV WVKANDRFQA RVLQKIGADH I IMPERDMG IRVARKMLDK RVLEFHPLGS GLAMTEFVVG SRLMGKTLSD ...String: MKTGDKQFAV IGLGRFGLAV CKELQDSGSQ VLAVDINEDR VKEAAGFVSQ AIVANCTHEE TVAELKLDD YDMVMIAIGA DVNASILATL IAKEAGVKSV WVKANDRFQA RVLQKIGADH I IMPERDMG IRVARKMLDK RVLEFHPLGS GLAMTEFVVG SRLMGKTLSD LALCKVEGVQ VL GYKRGPE IIKAPDMSTT LEIGDLIIVV GPQDKLANKL KSL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 5 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| ||||||||||||||||||
| Grid | Model: C-flat CF-MH-4C / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR | ||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 70 % / Chamber temperature: 283 K / Instrument: FEI VITROBOT MARK IV / Details: The sample was blotted for 10 seconds. |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 3200FSC |
|---|---|
| Electron beam | Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Calibrated defocus max: 2.5 µm / Calibrated defocus min: 1.5 µm / Calibrated magnification: 30675 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 4.2 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.5 µm / Nominal magnification: 20000 |
| Specialist optics | Energy filter - Name: In-column Omega Filter |
| Sample stage | Specimen holder model: JEOL 3200FSC CRYOHOLDER / Cooling holder cryogen: NITROGEN |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Frames/image: 2-40 / Number real images: 800 / Average exposure time: 8.0 sec. / Average electron dose: 27.0 e/Å2 |
-Image processing
| Particle selection | Number selected: 35400 |
|---|---|
| CTF correction | Software - Name: CTFFIND (ver. 3) |
| Startup model | Type of model: PDB ENTRY PDB model - PDB ID: |
| Initial angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) |
| Final 3D classification | Number classes: 3 / Avg.num./class: 12000 / Software - Name: RELION (ver. 1.4) |
| Final angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) |
| Final reconstruction | Number classes used: 1 / Applied symmetry - Point group: D3 (2x3 fold dihedral) / Resolution.type: BY AUTHOR / Resolution: 6.6 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 1.4) / Number images used: 20500 |
| FSC plot (resolution estimation) | 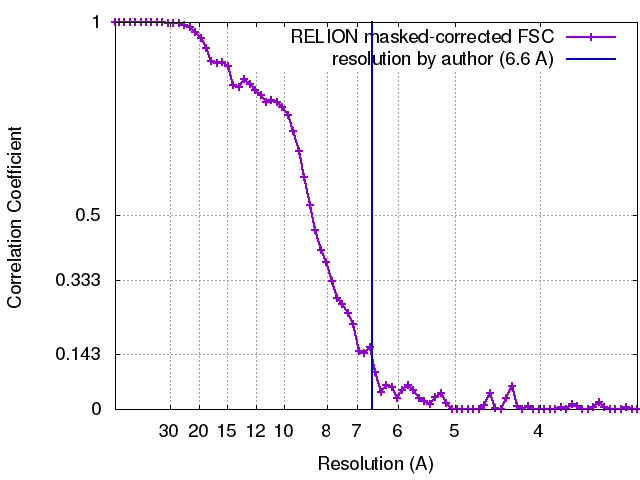 |

