- EMDB-8587: AAA ATPase Vps4 hexamer in closed conformation -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: EMDB / ID: EMD-8587
Title
AAA ATPase Vps4 hexamer in closed conformation
Map data
AAA ATPase Vps4 hexamer in closed conformation
Sample
Complex: Vps4
Protein or peptide: Vps4
Function / homology
Function and homology information
ESCRT IV complex / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / Sealing of the nuclear envelope (NE) by ESCRT-III / protein retention in Golgi apparatus / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / nuclear membrane reassembly / midbody abscission / vacuole organization ...ESCRT IV complex / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / Sealing of the nuclear envelope (NE) by ESCRT-III / protein retention in Golgi apparatus / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / nuclear membrane reassembly / midbody abscission / vacuole organization / multivesicular body sorting pathway / membrane fission / plasma membrane repair / late endosome to vacuole transport / multivesicular body assembly / reticulophagy / nucleus organization / endosomal transport / ATPase complex / autophagosome maturation / nuclear pore / macroautophagy / autophagy / protein transport / midbody / endosome / endoplasmic reticulum / ATP hydrolysis activity / protein homodimerization activity / ATP binding / membrane / identical protein binding / plasma membrane / cytoplasm Similarity search - Function
Vacuolar protein sorting-associated protein 4, MIT domain / MIT (microtubule interacting and transport) domain / MIT domain superfamily / Vps4 oligomerisation, C-terminal / MIT domain / Microtubule Interacting and Trafficking molecule domain / Vps4 C terminal oligomerisation domain / AAA ATPase, AAA+ lid domain / AAA+ lid domain / ATPase, AAA-type, conserved site ...Vacuolar protein sorting-associated protein 4, MIT domain / MIT (microtubule interacting and transport) domain / MIT domain superfamily / Vps4 oligomerisation, C-terminal / MIT domain / Microtubule Interacting and Trafficking molecule domain / Vps4 C terminal oligomerisation domain / AAA ATPase, AAA+ lid domain / AAA+ lid domain / ATPase, AAA-type, conserved site / AAA-protein family signature. / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01 GM095769
United States
National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Disease (NIH/NIDDK)
R01 DK090165
United States
Citation
Journal: Sci Adv / Year: 2017 Title: Mechanism of Vps4 hexamer function revealed by cryo-EM. Authors: Min Su / Emily Z Guo / Xinqiang Ding / Yan Li / Jeffrey T Tarrasch / Charles L Brooks / Zhaohui Xu / Georgios Skiniotis / Abstract: Vps4 is a member of AAA ATPase (adenosine triphosphatase associated with diverse cellular activities) that operates as an oligomer to disassemble ESCRT-III (endosomal sorting complex required for ...Vps4 is a member of AAA ATPase (adenosine triphosphatase associated with diverse cellular activities) that operates as an oligomer to disassemble ESCRT-III (endosomal sorting complex required for transport III) filaments, thereby catalyzing the final step in multiple ESCRT-dependent membrane remodeling events. We used electron cryo-microscopy to visualize oligomers of a hydrolysis-deficient Vps4 (vacuolar protein sorting-associated protein 4) mutant in the presence of adenosine 5'-triphosphate (ATP). We show that Vps4 subunits assemble into an asymmetric hexameric ring following an approximate helical path that sequentially stacks substrate-binding loops along the central pore. The hexamer is observed to adopt an open or closed ring configuration facilitated by major conformational changes in a single subunit. The structural transition of the mobile Vps4 subunit results in the repositioning of its substrate-binding loop from the top to the bottom of the central pore, with an associated translation of 33 Å. These structures, along with mutant-doping experiments and functional assays, provide evidence for a sequential and processive ATP hydrolysis mechanism by which Vps4 hexamers disassemble ESCRT-III filaments.
History
Deposition
Feb 3, 2017
-
Header (metadata) release
Mar 8, 2017
-
Map release
Nov 8, 2017
-
Update
Jan 29, 2020
-
Current status
Jan 29, 2020
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Function and homology information
Function and homology information ESCRT IV complex / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / Sealing of the nuclear envelope (NE) by ESCRT-III / protein retention in Golgi apparatus / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / nuclear membrane reassembly / midbody abscission / vacuole organization ...
ESCRT IV complex / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / Sealing of the nuclear envelope (NE) by ESCRT-III / protein retention in Golgi apparatus / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / nuclear membrane reassembly / midbody abscission / vacuole organization ...
 Authors
Authors United States, 2 items
United States, 2 items  Citation
Citation Structure visualization
Structure visualization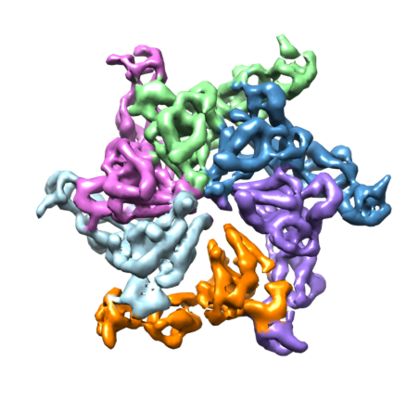
 Downloads & links
Downloads & links emd_8587.png
emd_8587.png http://ftp.pdbj.org/pub/emdb/structures/EMD-8587
http://ftp.pdbj.org/pub/emdb/structures/EMD-8587













 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy
