hemoglobin complex / oxygen carrier activity / oxygen binding / iron ion binding / heme binding / extracellular region / metal ion binding Similarity search - Function
Annelid erythrocruorin linker subunit, C-terminal / Erythrocruorin linker subunit, C-terminal superfamily / Extracellular hemoglobin linker subunit, heterodimerisation domain / Annelid erythrocruorin linker subunit C-terminus / Globin, extracellular / Erythrocruorin / Low-density lipoprotein receptor domain class A / Low-density lipoprotein (LDL) receptor class A, conserved site / LDL-receptor class A (LDLRA) domain signature. / LDL-receptor class A (LDLRA) domain profile. ...Annelid erythrocruorin linker subunit, C-terminal / Erythrocruorin linker subunit, C-terminal superfamily / Extracellular hemoglobin linker subunit, heterodimerisation domain / Annelid erythrocruorin linker subunit C-terminus / Globin, extracellular / Erythrocruorin / Low-density lipoprotein receptor domain class A / Low-density lipoprotein (LDL) receptor class A, conserved site / LDL-receptor class A (LDLRA) domain signature. / LDL-receptor class A (LDLRA) domain profile. / Myoglobin-like, M family globin domain / Low-density lipoprotein receptor domain class A / Low-density lipoprotein (LDL) receptor class A repeat / LDL receptor-like superfamily / Globin/Protoglobin / Globin family profile. / Globin / Globin / Globin-like superfamily Similarity search - Domain/homology
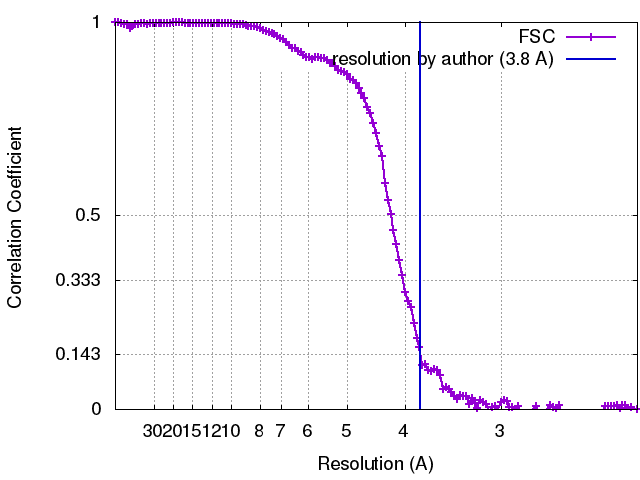
Journal: IUCrJ / Year: 2017 Title: Single-particle cryo-EM using alignment by classification (ABC): the structure of haemoglobin. Authors: Pavel Afanasyev / Charlotte Seer-Linnemayr / Raimond B G Ravelli / Rishi Matadeen / Sacha De Carlo / Bart Alewijnse / Rodrigo V Portugal / Navraj S Pannu / Michael Schatz / Marin van Heel / Abstract: Single-particle cryogenic electron microscopy (cryo-EM) can now yield near-atomic resolution structures of biological complexes. However, the reference-based alignment algorithms commonly used in ...Single-particle cryogenic electron microscopy (cryo-EM) can now yield near-atomic resolution structures of biological complexes. However, the reference-based alignment algorithms commonly used in cryo-EM suffer from reference bias, limiting their applicability (also known as the 'Einstein from random noise' problem). Low-dose cryo-EM therefore requires robust and objective approaches to reveal the structural information contained in the extremely noisy data, especially when dealing with small structures. A reference-free pipeline is presented for obtaining near-atomic resolution three-dimensional reconstructions from heterogeneous ('four-dimensional') cryo-EM data sets. The methodologies integrated in this pipeline include camera correction, movie-based full-data-set contrast transfer function determination, movie-alignment algorithms, (Fourier-space) multivariate statistical data compression and unsupervised classification, 'random-startup' three-dimensional reconstructions, four-dimensional structural refinements and Fourier shell correlation criteria for evaluating anisotropic resolution. The procedures exclusively use information emerging from the data set itself, without external 'starting models'. Euler-angle assignments are performed by angular reconstitution rather than by the inherently slower projection-matching approaches. The comprehensive 'ABC-4D' pipeline is based on the two-dimensional reference-free 'alignment by classification' (ABC) approach, where similar images in similar orientations are grouped by unsupervised classification. Some fundamental differences between X-ray crystallography single-particle cryo-EM data collection and data processing are discussed. The structure of the giant haemoglobin from at a global resolution of ∼3.8 Å is presented as an example of the use of the ABC-4D procedure.
History
Deposition
May 11, 2016
-
Header (metadata) release
Jun 15, 2016
-
Map release
Jul 26, 2017
-
Update
Jul 26, 2017
-
Current status
Jul 26, 2017
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Category: CCD / Film or detector model: FEI FALCON II (4k x 4k) / Number real images: 5235 / Average electron dose: 40 e/Å2 / Bits/pixel: 16
Experimental equipment
Model: Titan Krios / Image courtesy: FEI Company
-
Image processing
CTF correction
Details: Phase flipping of micrograph patches (ctf2d-find)
Final two d classification
Number classes: 85000
Final angle assignment
Details: Imagic D6 asymmetric triangle: 0
Final reconstruction
Applied symmetry - Point group: D6 (2x6 fold dihedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 3.8 Å / Resolution method: OTHER / Software - Name: Imagic-4D / Number images used: 85000
Details
Reference-free Alignment By Classification (ABC-4D). Camera correction, CTF determination, particle-picking, MSA unsupervised classification, 3D reconstruction, all in IMAGIC-4D.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Lumbricus terrestris /
Lumbricus terrestris /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation




 Structure visualization
Structure visualization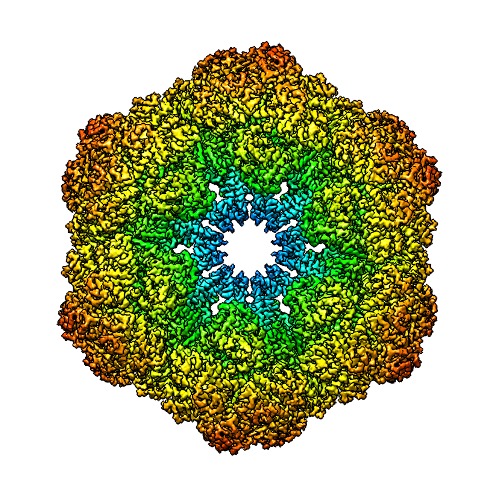
 Downloads & links
Downloads & links http://ftp.pdbj.org/pub/emdb/structures/EMD-3434
http://ftp.pdbj.org/pub/emdb/structures/EMD-3434














 Z
Z Y
Y X
X







 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy