 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3089: Structural basis for DNA strand separation by a hexameric replica... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3089 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structural basis for DNA strand separation by a hexameric replicative helicase | |||||||||
 Map data Map data | The 6-fold symmetrized structure of the full-length E1 helicase | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords |  papillomavirus / helicase / DNA replication fork / electron microscopy / structural analysis papillomavirus / helicase / DNA replication fork / electron microscopy / structural analysis | |||||||||
| Function / homology |  Function and homology informationDNA helicase activity / DNA helicase / DNA replication / host cell nucleus / ATP hydrolysis activity / DNA binding / ATP binding Function and homology informationDNA helicase activity / DNA helicase / DNA replication / host cell nucleus / ATP hydrolysis activity / DNA binding / ATP bindingSimilarity search - Function | |||||||||
| Biological species |  Bovine papillomavirus Bovine papillomavirus | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 18.0 Å | |||||||||
 Authors Authors | Chaban Y / Stead JA / Ryzhenkova K / Whelan F / Lamber K / Antson A / Sanders CM / Orlova EV | |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2015 Title: Structural basis for DNA strand separation by a hexameric replicative helicase. Authors: Yuriy Chaban / Jonathan A Stead / Ksenia Ryzhenkova / Fiona Whelan / Ekaterina P Lamber / Alfred Antson / Cyril M Sanders / Elena V Orlova /  Abstract: Hexameric helicases are processive DNA unwinding machines but how they engage with a replication fork during unwinding is unknown. Using electron microscopy and single particle analysis we determined ...Hexameric helicases are processive DNA unwinding machines but how they engage with a replication fork during unwinding is unknown. Using electron microscopy and single particle analysis we determined structures of the intact hexameric helicase E1 from papillomavirus and two complexes of E1 bound to a DNA replication fork end-labelled with protein tags. By labelling a DNA replication fork with streptavidin (dsDNA end) and Fab (5' ssDNA) we located the positions of these labels on the helicase surface, showing that at least 10 bp of dsDNA enter the E1 helicase via a side tunnel. In the currently accepted 'steric exclusion' model for dsDNA unwinding, the active 3' ssDNA strand is pulled through a central tunnel of the helicase motor domain as the dsDNA strands are wedged apart outside the protein assembly. Our structural observations together with nuclease footprinting assays indicate otherwise: strand separation is taking place inside E1 in a chamber above the helicase domain and the 5' passive ssDNA strands exits the assembly through a separate tunnel opposite to the dsDNA entry point. Our data therefore suggest an alternative to the current general model for DNA unwinding by hexameric helicases. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3089.map.gz | 1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3089-v30.xmlemd-3089.xml | 13.3 KB 13.3 KB | Display Display | EMDB header |
| Images | 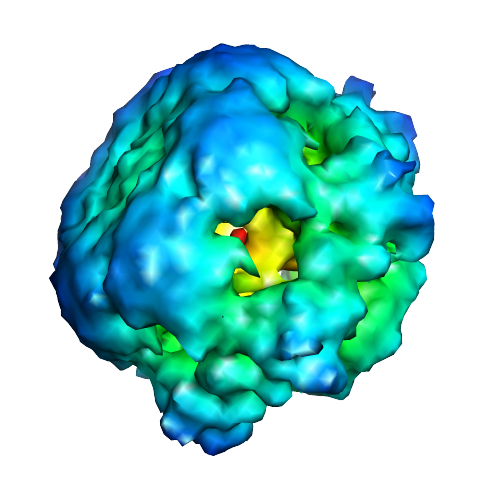 image3089.png image3089.png | 164.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3089ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3089 http://ftp.pdbj.org/pub/emdb/structures/EMD-3089ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3089 | HTTPS FTP |
-Related structure data










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_3089.map.gz / Format: CCP4 / Size: 29.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | The 6-fold symmetrized structure of the full-length E1 helicase | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.6 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
-Supplemental data
- Sample components
Sample components
-Entire : Full-length E1 helicase from Bovine Papillomavirus bound to ssDNA T30
| Entire | Name: Full-length E1 helicase from Bovine Papillomavirus bound to ssDNA T30 |
|---|---|
| Components |
|
-Supramolecule #1000: Full-length E1 helicase from Bovine Papillomavirus bound to ssDNA T30
| Supramolecule | Name: Full-length E1 helicase from Bovine Papillomavirus bound to ssDNA T30 type: sample / ID: 1000 / Oligomeric state: hexamer / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 410 KDa / Theoretical: 410 KDa / Method: Theoretical calculation |
-Macromolecule #1: Full-length hexameric E1 helicase
| Macromolecule | Name: Full-length hexameric E1 helicase / type: protein_or_peptide / ID: 1 / Name.synonym: FLE1 / Number of copies: 1 / Oligomeric state: hexamer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: Bovine papillomavirus / synonym: Bovine papillomavirus / Organelle: nucleus / Location in cell: nucleus |
| Molecular weight | Experimental: 410 KDa / Theoretical: 410 KDa |
| Recombinant expression | Organism:  Escherichia coli (E. coli) / Recombinant strain: BL21(DE3) / Recombinant plasmid: pET11c Escherichia coli (E. coli) / Recombinant strain: BL21(DE3) / Recombinant plasmid: pET11c |
| Sequence | UniProtKB: Replication protein E1 |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3 mg/mL |
|---|---|
| Buffer | pH: 8 Details: 10 mM Tris-Cl pH 8.0, 225 mM NaCl,2 mM DTT, 0.1 mM PMSF, 0.1 mM EDTA |
| Staining | Type: NEGATIVE / Details: Sample was stained with 2% uranyl acetate |
| Grid | Details: Sample was applied on to carbon-coated copper grids (400 mesh, freshly glow-discharged in air) |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Electron beam | Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 67000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELDBright-field microscopy / Cs: 2.1 mm / Nominal defocus max: 1.5 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 62000 |
| Sample stage | Specimen holder: Negative stain holder / Specimen holder model: OTHER |
| Temperature | Min: 291 K / Max: 296 K / Average: 293 K |
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at 100,000 times magnification |
| Date | Apr 15, 2012 |
| Image recording | Category: CCD / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Digitization - Sampling interval: 1.6 µm / Number real images: 45 / Average electron dose: 20 e/Å2 / Camera length: 1000 / Details: No subframe averaging was used. / Bits/pixel: 16 |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| CTF correction | Details: frames |
|---|---|
| Final two d classification | Number classes: 500 |
| Final angle assignment | Details: Angular reconstitution, 6-fold symmetry |
| Final reconstruction | Applied symmetry - Point group: C6 (6 fold cyclic) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 18.0 Å / Resolution method: OTHER / Software - Name: Imagic, CTFit, CTFFIND3 / Details: Final map was calculated from 500 classes / Number images used: 3500 |
| Details | Particle picking was carried out automatically using BOXER software. Initial references were prepared using several manually selected protein complex images representing different views. |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C / Chain - #3 - Chain ID: D / Chain - #4 - Chain ID: E / Chain - #5 - Chain ID: F |
|---|---|
| Software | Name: Chimera |
| Details | The domains were separately fitted by manual docking using Chimera |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: correlation coefficient |

-Atomic model buiding 2
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Software | Name: Chimera |
| Details | The domains were separately fitted by manual docking using Chimera |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: correlation coefficient |

