 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1n83: Crystal Structure of the complex between the Orphan Nuclear Hormo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n83 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the complex between the Orphan Nuclear Hormone Receptor ROR(alpha)-LBD and Cholesterol | ||||||
 Components Components | Nuclear receptor ROR-alpha | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / three-layered alpha helical sandwich /  receptor / transcription regulation / nuclear protein / DNA binding receptor / transcription regulation / nuclear protein / DNA binding | ||||||
| Function / homology |  Function and homology information Function and homology informationcGMP metabolic process / cerebellar Purkinje cell differentiation / muscle cell differentiation / cerebellar granule cell precursor proliferation / T-helper 17 cell differentiation / ligand-activated transcription factor activity / cellular response to sterol / regulation of steroid metabolic process / triglyceride homeostasis / intracellular receptor signaling pathway ...cGMP metabolic process / cerebellar Purkinje cell differentiation / muscle cell differentiation / cerebellar granule cell precursor proliferation / T-helper 17 cell differentiation / ligand-activated transcription factor activity / cellular response to sterol / regulation of steroid metabolic process / triglyceride homeostasis / intracellular receptor signaling pathway / positive regulation of circadian rhythm / oxysterol binding / regulation of smoothened signaling pathway / regulation of macrophage activation / negative regulation of fat cell differentiation / regulation of glucose metabolic process / positive regulation of vascular endothelial growth factor production / cellular response to interleukin-1 / negative regulation of canonical NF-kappaB signal transduction / RORA activates gene expression / nitric oxide biosynthetic process / xenobiotic metabolic process / cholesterol homeostasis / transcription corepressor binding / transcription coregulator binding / RNA polymerase II transcription regulatory region sequence-specific DNA binding / circadian regulation of gene expression / SUMOylation of intracellular receptors / Heme signaling / transcription coactivator binding / PPARA activates gene expression / negative regulation of inflammatory response / beta-catenin binding / Nuclear Receptor transcription pathway / nuclear receptor activity / Circadian Clock / cellular response to tumor necrosis factor / cellular response to hypoxia / angiogenesis / Interleukin-4 and Interleukin-13 signaling / sequence-specific DNA binding / DNA-binding transcription factor activity, RNA polymerase II-specific / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / chromatin / nucleolus / regulation of DNA-templated transcription / regulation of transcription by RNA polymerase II / positive regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / DNA binding / zinc ion binding / nucleoplasm / nucleusSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / SIRAS / Resolution: 1.63 Å | ||||||
 Authors Authors | Kallen, J.A. / Schlaeppi, J.M. / Bitsch, F. / Geisse, S. / Geiser, M. / Delhon, I. / Fournier, B. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: X-ray Structure of hROR(alpha) LBD at 1.63A: Structural and Functional data that Cholesterol or a Cholesterol derivative is the natural ligand of ROR(alpha) Authors: Kallen, J.A. / Schlaeppi, J.M. / Bitsch, F. / Geisse, S. / Geiser, M. / Delhon, I. / Fournier, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n83.cif.gz | 75.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n83.ent.gz | 54.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1n83.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n8/1n83ftp://data.pdbj.org/pub/pdb/validation_reports/n8/1n83 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31514.279 Da / Num. of mol.: 1 / Fragment: Ligand Binding Domain, residues 304-556 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:   Spodoptera frugiperda (fall armyworm) / Strain (production host): Sf9 / References: UniProt: P35398 Spodoptera frugiperda (fall armyworm) / Strain (production host): Sf9 / References: UniProt: P35398 |
|---|---|
| #2: Chemical | ChemComp-CLR / Cholesterol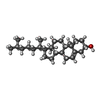  Mass: 386.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H46O Mass: 386.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H46O |
| #3: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 419 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 46.5 % | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: PEG 4000, sodium acetate, hepes, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 8.4 | ||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 105 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM1A / Wavelength: 0.8727 Å / Beamline: BM1A / Wavelength: 0.8727 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 30, 2001 |
| Radiation | Monochromator: Si Channel / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8727 Å / Relative weight: 1 |
| Reflection | Resolution: 1.63→15 Å / Num. all: 38783 / Num. obs: 38783 / % possible obs: 95.36 % / Observed criterion σ(I): 0 / Redundancy: 3.65 % / Biso Wilson estimate: 22.1 Å2 / Rsym value: 0.04 / Net I/σ(I): 32 |
| Reflection shell | Resolution: 1.63→1.69 Å / Redundancy: 2.5 % / Mean I/σ(I) obs: 2.1 / Num. unique all: 2775 / Rsym value: 0.433 / % possible all: 67.3 |
| Reflection | *PLUS Num. measured all: 141730 / Rmerge(I) obs: 0.04 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SIRAS / Resolution: 1.63→15 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.945 / SU B: 2.74 / SU ML: 0.095 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.104 / ESU R Free: 0.098 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.739 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.63→15 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.63→1.672 Å / Total num. of bins used: 20 /
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 15 Å / Num. reflection obs: 36570 / Num. reflection Rfree: 1907 / % reflection Rfree: 5 % / Rfactor Rfree: 0.23 / Rfactor Rwork: 0.199 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
