 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8yle: Crystal structure of Werner syndrome helicase complexed with AMP-PCP -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8yle | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Werner syndrome helicase complexed with AMP-PCP | ||||||
 Components Components | Bifunctional 3'-5' exonuclease/ATP-dependent helicase WRN | ||||||
 Keywords Keywords |  HYDROLASE / DNA repair helicase HYDROLASE / DNA repair helicase | ||||||
| Function / homology |  Function and homology information Function and homology information3'-flap-structured DNA binding / positive regulation of strand invasion / telomeric G-quadruplex DNA binding / forked DNA-dependent helicase activity / positive regulation of hydrolase activity / 8-hydroxy-2'-deoxyguanosine DNA binding / telomeric D-loop binding / regulation of growth rate / telomere maintenance via semi-conservative replication / G-quadruplex DNA unwinding ...3'-flap-structured DNA binding / positive regulation of strand invasion / telomeric G-quadruplex DNA binding / forked DNA-dependent helicase activity / positive regulation of hydrolase activity / 8-hydroxy-2'-deoxyguanosine DNA binding / telomeric D-loop binding / regulation of growth rate / telomere maintenance via semi-conservative replication / G-quadruplex DNA unwinding / t-circle formation / telomeric D-loop disassembly / Y-form DNA binding / DNA 3'-5' helicase / four-way junction helicase activity / G-quadruplex DNA binding / MutLalpha complex binding / bubble DNA binding / Processive synthesis on the C-strand of the telomere / Impaired BRCA2 binding to PALB2 / Removal of the Flap Intermediate from the C-strand / protein localization to nucleolus / Defective homologous recombination repair (HRR) due to BRCA1 loss of function / Defective HDR through Homologous Recombination Repair (HRR) due to PALB2 loss of BRCA1 binding function / Defective HDR through Homologous Recombination Repair (HRR) due to PALB2 loss of BRCA2/RAD51/RAD51C binding function / Homologous DNA Pairing and Strand Exchange / Resolution of D-loop Structures through Synthesis-Dependent Strand Annealing (SDSA) / response to UV-C / Resolution of D-loop Structures through Holliday Junction Intermediates / exonuclease activity / DNA duplex unwinding / HDR through Single Strand Annealing (SSA) / Impaired BRCA2 binding to RAD51 / DNA metabolic process / 3'-5' DNA helicase activity / DNA synthesis involved in DNA repair / replication fork processing / DNA unwinding involved in DNA replication / Presynaptic phase of homologous DNA pairing and strand exchange / replicative senescence / SUMOylation of DNA damage response and repair proteins / four-way junction DNA binding / isomerase activity / 3'-5' exonuclease activity / cellular response to starvation / DNA helicase activity / telomere maintenance / replication fork / determination of adult lifespan / double-strand break repair via homologous recombination / base-excision repair / HDR through Homologous Recombination (HRR) / G2/M DNA damage checkpoint / cellular response to gamma radiation / cellular senescence / double-strand break repair / chromosome / manganese ion binding / Processing of DNA double-strand break ends / response to oxidative stress / Regulation of TP53 Activity through Phosphorylation / DNA replication / chromosome, telomeric region / Hydrolases; Acting on ester bonds / nuclear speck / centrosome / DNA damage response / chromatin binding / protein-containing complex binding / nucleolus / magnesium ion binding / protein homodimerization activity / ATP hydrolysis activity / DNA binding / nucleoplasm / ATP binding / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.86 Å | ||||||
 Authors Authors | Yang, Y. / Fu, L. / Sun, X. / Cheng, H. / Chen, R. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Structure of werner syndrome helicase complexed with AMP-PCP at 1.86 Angstroms resolution. Authors: Chen, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8yle.cif.gz | 219.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8yle.ent.gz | 142.4 KB | Display | PDB format |
| PDBx/mmJSON format | 8yle.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yl/8yleftp://data.pdbj.org/pub/pdb/validation_reports/yl/8yle | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|
-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 49003.477 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: WRN, RECQ3, RECQL2 / Production host:  Escherichia coli (E. coli) Escherichia coli (E. coli)References: UniProt: Q14191, Hydrolases; Acting on ester bonds, DNA helicase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ACP / 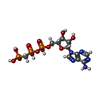  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Feature type: SUBJECT OF INVESTIGATION / Comment: AMP-PCP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Feature type: SUBJECT OF INVESTIGATION / Comment: AMP-PCP, energy-carrying molecule analogue*YM | ||||||
| #3: Chemical | Ethylene glycol  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 235 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 235 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.85 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion / pH: 6.5 Details: pH 6.5,0.1 M Amino acids,0.1 M Buffer System 1, 30.00% % v/v EDO_P8K |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL02U1 / Wavelength: 0.979183 Å / Beamline: BL02U1 / Wavelength: 0.979183 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Jun 29, 2023 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979183 Å / Relative weight: 1 |
| Reflection | Resolution: 1.86→30.05 Å / Num. obs: 38310 / % possible obs: 98.3 % / Redundancy: 4.3 % / Biso Wilson estimate: 22.22 Å2 / CC1/2: 0.993 / CC star: 0.998 / Rmerge(I) obs: 0.139 / Rpim(I) all: 0.07505 / Rrim(I) all: 0.1585 / Net I/σ(I): 8.28 |
| Reflection shell | Resolution: 1.86→1.926 Å / Redundancy: 4.4 % / Rmerge(I) obs: 1.123 / Mean I/σ(I) obs: 2.23 / Num. unique obs: 3782 / CC1/2: 0.631 / CC star: 0.88 / % possible all: 97.54 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.86→30.05 Å / SU ML: 0.2025 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 21.8709 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.67 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.86→30.05 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Refine-ID: X-RAY DIFFRACTION / Auth asym-ID: A / Label asym-ID: A
|
