 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7uxs: Crystal structure of the BcThsA SLOG domain in complex with 3'cADPR -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7uxs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the BcThsA SLOG domain in complex with 3'cADPR | |||||||||
 Components Components | BcThsA | |||||||||
 Keywords Keywords |  HYDROLASE / cADPR isomer / bacterial TIR / Thoeris defense system / ThsA / ThsB / antiphage defense / NAD+ HYDROLASE / cADPR isomer / bacterial TIR / Thoeris defense system / ThsA / ThsB / antiphage defense / NAD+ | |||||||||
| Function / homology | Chem-OJC Function and homology information Function and homology information | |||||||||
| Biological species |  Bacillus subtilis (bacteria) Bacillus subtilis (bacteria) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.57 Å | |||||||||
 Authors Authors | Shi, Y. / Masic, V. / Mosaiab, T. / Ve, T. | |||||||||
| Funding support |  Australia, 2items Australia, 2items
| |||||||||
 Citation Citation | Journal: Science / Year: 2022 Title: Cyclic ADP ribose isomers: Production, chemical structures, and immune signaling. Authors: Mohammad K Manik / Yun Shi / Sulin Li / Mark A Zaydman / Neha Damaraju / Samuel Eastman / Thomas G Smith / Weixi Gu / Veronika Masic / Tamim Mosaiab / James S Weagley / Steven J Hancock / ...Authors: Mohammad K Manik / Yun Shi / Sulin Li / Mark A Zaydman / Neha Damaraju / Samuel Eastman / Thomas G Smith / Weixi Gu / Veronika Masic / Tamim Mosaiab / James S Weagley / Steven J Hancock / Eduardo Vasquez / Lauren Hartley-Tassell / Nestoras Kargios / Natsumi Maruta / Bryan Y J Lim / Hayden Burdett / Michael J Landsberg / Mark A Schembri / Ivan Prokes / Lijiang Song / Murray Grant / Aaron DiAntonio / Jeffrey D Nanson / Ming Guo / Jeffrey Milbrandt / Thomas Ve / Bostjan Kobe /   Abstract: Cyclic adenosine diphosphate (ADP)-ribose (cADPR) isomers are signaling molecules produced by bacterial and plant Toll/interleukin-1 receptor (TIR) domains via nicotinamide adenine dinucleotide ...Cyclic adenosine diphosphate (ADP)-ribose (cADPR) isomers are signaling molecules produced by bacterial and plant Toll/interleukin-1 receptor (TIR) domains via nicotinamide adenine dinucleotide (oxidized form) (NAD) hydrolysis. We show that v-cADPR (2'cADPR) and v2-cADPR (3'cADPR) isomers are cyclized by O-glycosidic bond formation between the ribose moieties in ADPR. Structures of 2'cADPR-producing TIR domains reveal conformational changes that lead to an active assembly that resembles those of Toll-like receptor adaptor TIR domains. Mutagenesis reveals a conserved tryptophan that is essential for cyclization. We show that 3'cADPR is an activator of ThsA effector proteins from the bacterial antiphage defense system termed Thoeris and a suppressor of plant immunity when produced by the effector HopAM1. Collectively, our results reveal the molecular basis of cADPR isomer production and establish 3'cADPR in bacteria as an antiviral and plant immunity-suppressing signaling molecule. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7uxs.cif.gz | 207.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7uxs.ent.gz | 134.2 KB | Display | PDB format |
| PDBx/mmJSON format | 7uxs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ux/7uxsftp://data.pdbj.org/pub/pdb/validation_reports/ux/7uxs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7uwgC  7uxrC  7uxtC  7uxuC  6lhxS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22350.076 Da / Num. of mol.: 2 / Fragment: SLOG domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacillus subtilis (bacteria) / Production host: Escherichia coli (E. coli)#2: Chemical | 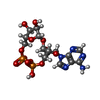  Mass: 541.300 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H21N5O13P2 / Feature type: SUBJECT OF INVESTIGATION Mass: 541.300 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H21N5O13P2 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | Glycerol  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-SO4 / | Sulfate  Mass: 96.063 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: SO4#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 365 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 365 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.22 Å3/Da / Density % sol: 44.52 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop Details: 0.1 M Bis-Tris, pH 5.5, 0.1 M ammonium sulfate, 25-29% PEG3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron / Beamline: MX2 / Wavelength: 0.9537 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Feb 17, 2022 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9537 Å / Relative weight: 1 |
| Reflection | Resolution: 1.57→46.16 Å / Num. obs: 56724 / % possible obs: 99.7 % / Redundancy: 6.6 % / Biso Wilson estimate: 15.42 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.038 / Rpim(I) all: 0.016 / Rrim(I) all: 0.042 / Net I/σ(I): 20.2 |
| Reflection shell | Resolution: 1.57→1.59 Å / Redundancy: 6.4 % / Rmerge(I) obs: 0.242 / Mean I/σ(I) obs: 4.6 / Num. unique obs: 2652 / CC1/2: 0.97 / Rpim(I) all: 0.102 / Rrim(I) all: 0.264 / % possible all: 95.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 6LHX Resolution: 1.57→36.87 Å / SU ML: 0.1435 / Cross valid method: FREE R-VALUE / σ(F): 1.39 / Phase error: 15.2414 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.84 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.57→36.87 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
