 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7u9i: Co-crystal structure of human CARM1 in complex with MT556 inhibitor -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7u9i | ||||||
|---|---|---|---|---|---|---|---|
| Title | Co-crystal structure of human CARM1 in complex with MT556 inhibitor | ||||||
 Components Components | Histone-arginine methyltransferase CARM1 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / MTase / Histone arginine methyltransferase / PRMT4 /  CARM1 / SGC / Structural Genomics / Structural Genomics Consortium / TRANSFERASE-TRANSFERASE INHIBITOR complex CARM1 / SGC / Structural Genomics / Structural Genomics Consortium / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology information: / histone H3R17 methyltransferase activity / histone H3R2 methyltransferase activity / negative regulation of dendrite development / type I protein arginine methyltransferase / protein-arginine omega-N asymmetric methyltransferase activity / protein methyltransferase activity / histone arginine N-methyltransferase activity / regulation of intracellular estrogen receptor signaling pathway / replication fork reversal ...: / histone H3R17 methyltransferase activity / histone H3R2 methyltransferase activity / negative regulation of dendrite development / type I protein arginine methyltransferase / protein-arginine omega-N asymmetric methyltransferase activity / protein methyltransferase activity / histone arginine N-methyltransferase activity / regulation of intracellular estrogen receptor signaling pathway / replication fork reversal / protein-arginine N-methyltransferase activity / positive regulation of epithelial cell apoptotic process / histone methyltransferase activity / positive regulation of transcription by RNA polymerase I / nuclear replication fork / positive regulation of fat cell differentiation / response to cAMP / RORA activates gene expression / Regulation of lipid metabolism by PPARalpha / TP53 Regulates Transcription of Genes Involved in G2 Cell Cycle Arrest / BMAL1:CLOCK,NPAS2 activates circadian gene expression / Activation of gene expression by SREBF (SREBP) / nuclear receptor coactivator activity / lysine-acetylated histone binding / Heme signaling / Transcriptional activation of mitochondrial biogenesis / PPARA activates gene expression / Cytoprotection by HMOX1 / beta-catenin binding / RMTs methylate histone arginines / Transcriptional regulation of white adipocyte differentiation / Circadian Clock / DNA-binding transcription factor binding / Estrogen-dependent gene expression / transcription coactivator activity / transcription cis-regulatory region binding / positive regulation of cell population proliferation / regulation of DNA-templated transcription / nucleoplasm / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å | ||||||
 Authors Authors | Zeng, H. / Perveen, S. / Dong, A. / Hutchinson, A. / Seitova, A. / Gibson, E. / Hajian, T. / Li, Y. / Gao, Y.D. / Schneider, S. ...Zeng, H. / Perveen, S. / Dong, A. / Hutchinson, A. / Seitova, A. / Gibson, E. / Hajian, T. / Li, Y. / Gao, Y.D. / Schneider, S. / Siliphaivanh, P. / Sloman, D. / Nicholson, B. / Fischer, C. / Hicks, J. / Vedadi, M. / Brown, P.J. / Arrowsmith, C.H. / Edwards, A.M. / Halabelian, L. / Structural Genomics Consortium (SGC) | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Co-crystal structure of human CARM1 in complex with MT556 inhibitor Authors: Zeng, H. / Perveen, S. / Dong, A. / Hutchinson, A. / Seitova, A. / Gibson, E. / Hajian, T. / Li, Y. / Gao, Y.D. / Schneider, S. / Siliphaivanh, P. / Sloman, D. / Nicholson, B. / Fischer, C. ...Authors: Zeng, H. / Perveen, S. / Dong, A. / Hutchinson, A. / Seitova, A. / Gibson, E. / Hajian, T. / Li, Y. / Gao, Y.D. / Schneider, S. / Siliphaivanh, P. / Sloman, D. / Nicholson, B. / Fischer, C. / Hicks, J. / Vedadi, M. / Brown, P.J. / Arrowsmith, C.H. / Edwards, A.M. / Halabelian, L. / Structural Genomics Consortium (SGC) | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7u9i.cif.gz | 149.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7u9i.ent.gz | 113.8 KB | Display | PDB format |
| PDBx/mmJSON format | 7u9i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/u9/7u9iftp://data.pdbj.org/pub/pdb/validation_reports/u9/7u9i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5dxjS S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39033.500 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CARM1, PRMT4 / Plasmid: pFBOH-MHL / Cell line (production host): Sf9 / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: Q86X55, type I protein arginine methyltransferase #2: Chemical | 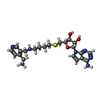  Mass: 472.604 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H32N6O3S / Feature type: SUBJECT OF INVESTIGATION Mass: 472.604 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H32N6O3S / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-UNX /   Num. of mol.: 14 / Source method: obtained synthetically Num. of mol.: 14 / Source method: obtained synthetically#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 209 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 209 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 49.06 % / Mosaicity: 0.35 ° |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / Details: 20% PEG3350, 0.2M di-Sodium Tartrate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E SUPERBRIGHT / Wavelength: 1.54 Å | ||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU SATURN A200 / Detector: CCD / Date: Jan 21, 2020 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→44.35 Å / Num. obs: 49481 / % possible obs: 97 % / Redundancy: 5.2 % / CC1/2: 0.998 / Rmerge(I) obs: 0.087 / Rpim(I) all: 0.04 / Rrim(I) all: 0.096 / Net I/σ(I): 13.7 / Num. measured all: 258148 / Scaling rejects: 114 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5DXJ Resolution: 2→40.21 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.919 / SU B: 5.383 / SU ML: 0.146 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.196 / ESU R Free: 0.188 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 101.34 Å2 / Biso mean: 30.477 Å2 / Biso min: 14.93 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→40.21 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
