 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7b9p: Structure of Ribonucleotide reductase from Rhodobacter sphaeroides -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7b9p | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Ribonucleotide reductase from Rhodobacter sphaeroides | ||||||
 Components Components | Vitamin B12-dependent ribonucleotide reductase | ||||||
 Keywords Keywords |  OXIDOREDUCTASE / Ribonucleotide Reductase / Thiyl Radical Enzyme / Allosteric Effector OXIDOREDUCTASE / Ribonucleotide Reductase / Thiyl Radical Enzyme / Allosteric Effector | ||||||
| Function / homology |  Function and homology informationcobalamin binding / ribonucleoside-diphosphate reductase / ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor / cobalt ion binding / DNA biosynthetic process / DNA replication / nucleotide binding Function and homology informationcobalamin binding / ribonucleoside-diphosphate reductase / ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor / cobalt ion binding / DNA biosynthetic process / DNA replication / nucleotide bindingSimilarity search - Function | ||||||
| Biological species |  Rhodobacter sphaeroides (bacteria) Rhodobacter sphaeroides (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.646 Å | ||||||
 Authors Authors | Wilk, P. / Feiler, C. / Loderer, C. / Kabinger, F. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2022 Title: HUG Domain Is Responsible for Active Dimer Stabilization in an NrdJd Ribonucleotide Reductase. Authors: Fietze, T. / Wilk, P. / Kabinger, F. / Anoosheh, S. / Hofer, A. / Lundin, D. / Feiler, C.G. / Weiss, M.S. / Loderer, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7b9p.cif.gz | 367.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7b9p.ent.gz | 302.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7b9p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b9/7b9pftp://data.pdbj.org/pub/pdb/validation_reports/b9/7b9p | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 100522.297 Da / Num. of mol.: 1 / Mutation: V926Stop Source method: isolated from a genetically manipulated source Details: From the full length protein with 1218 aa, the C-terminal CRD domain was deleted by insertion of a stop codon at postion 926. Source: (gene. exp.) Rhodobacter sphaeroides (strain ATCC 17023 / 2.4.1 / NCIB 8253 / DSM 158) (bacteria)Strain: ATCC 17023 / 2.4.1 / NCIB 8253 / DSM 158 / Gene: nrd, RSP_2495 / Plasmid: pET28b(+) / Details (production host): N-Terminal His-Tag fusion / Production host: Escherichia coli BL21(DE3) (bacteria)References: UniProt: Q3J3H6, ribonucleoside-diphosphate reductase |
|---|---|
| #2: Chemical | ChemComp-DTP / Deoxyadenosine triphosphate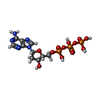  Mass: 491.182 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O12P3 Mass: 491.182 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O12P3 |
| #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: Mg |
| #4: Water | ChemComp-HOH / Water Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.18 Å3/Da / Density % sol: 76.26 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 8 Details: Precipitant: 30% PEG 1500, 20% Glycerol Protein solution: 45 mg/mL Protein + 100 uM dATP Hanging drop experiment: 1.0 uL Protein solution + 1.0 uL Precipitant over 500 uL precipitant |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 Å / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jan 26, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 2.646→47.86 Å / Num. obs: 62949 / % possible obs: 99.83 % / Redundancy: 19.29 % / CC1/2: 0.999 / Rmerge(I) obs: 0.169 / Rpim(I) all: 0.04 / Rrim(I) all: 0.174 / Net I/σ(I): 13.9 |
| Reflection shell | Resolution: 2.65→2.74 Å / Redundancy: 19.7 % / Rmerge(I) obs: 7.684 / Mean I/σ(I) obs: 0.3 / Num. unique obs: 6078 / CC1/2: 0.148 / Rpim(I) all: 1.756 / Rrim(I) all: 7.886 / % possible all: 98.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: ensemble Resolution: 2.646→47.86 Å / SU ML: 0.54 / Cross valid method: THROUGHOUT / σ(F): 1.33 / Phase error: 32.05 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 230 Å2 / Biso mean: 114.4113 Å2 / Biso min: 58.67 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.646→47.86 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
